Integrals associated with the Potts model
Victor S. Adamchik
Computer Science, Carnegie Mellon University,5000 Forbes Av.,
Pittsburgh, 15213 PA, USA
In this talk I will discuss an
approach to analytic evaluation of integrals associated with the Potts model.
The results are given in terms of the Barnes and multiple γ functions.
Graph-theoretic approach to characterization
of proteome maps
Željko Bajzer1 and Milan Randić2,3
1Department of Biochemistry and Molecular
Biology, Mayo Clinic College of Medicine, Rochester, MN 55905, USA
2Department of Mathematics and Computer
Science, Drake University, Des Moines, IA 50010, USA
3National Institute of Chemistry, Hajdrihova
19, 1000 Ljubljana, Slovenia
Standard bioinformatics methods for data reduction in proteomics are designed to emphasize changes in protein expression as a function of experimental perturbation. These
methods still cannot reveal how the changes in expression levels of numerous proteins in a system are interconnected. Graph-theoretical methods used in computational
chemistry offer the possibility to describe interconnectedness of levels of proteins by associating graphs to 2-D proteome maps and describing the underlying patterns by
graphs invariants. In particular we consider here graphs obtained by connecting all of the spots on the map within chose critical distance and graphs in which spots are
connected to their closest neighbours. The graphs are further numerically represented by adjacency matrix, Euclidean distance matrix and D/D matrix, and these are
characterized by invariants such as the leading eigenvalue and average sum of rows. Using actual data for proteome maps related to influence of several perxisome
proliferators to mouse liver cells, we show that our approach leads to characterization of proteome maps capable of revealing hidden patterns. This is accomplished by
comparison to results obtained from synthetic proteome maps with randomly generated spots.
Granulometric roundness parameter determined on model and natural sediment grains,
suggested as
a possible sphericity index
Yoganand Balagurunathan1 and Stanislav Frančišković-Bilinski2
1Translational Genomics Research Institute,
Phoenix, AZ 85004, USA
2Ruđer Bošković Institute, HR-10002
Zagreb, POB 180, Croatia
Present research in progress is a continuation of our recent studies on morphological granulometric analysis of sediment images1 and of real siliceous and calcareous estuarine
and marine sediments. 2 The first preliminary attempt to compare granulometric roundness with Wadell’s sphericity and roundness was reported on very few simulated and
real imaged calcareous marine sediment grains. 3
Digital granulometries has been traditionally used for shape analysis and its size distributions or the pattern spectrum and its moments form good shape descriptors. These
features find applications in shape characterization, classification and parameter estimation. We propose to use granulometric roundness parameter, which is defined as the
ratio of pattern spectrum variability to its mean all with respect to a circular reference shape (structuring element). As the shapes deviates from circularity the pattern
spectrum shows a larger spread in its density function and the amount of spread is related to the deviation from perfect circular shape. These measures, along with
conventional shape features, could be latter used to predict the conventional measures.
In our current work we present results of proposed granulometric roundness measure, compared with the conventional Wadell’s measures. This comparison is carried out
for few simulated and real imaged grain samples taken along Soča River (Slovenia). The digital methods for granulometric roundness measure are faster and accurate. They
can be suggested as possible sphericity index in sedimentary research.
1. Y. Balagurunathan, E.R. Dougherty,
S. Frančišković-Bilinski, H. Bilinski, N. Vdović (2001)
Morphological granulometric analysis of sediment images. Image Analysis and Stereology 20:
87-99.
2. S. Frančišković-Bilinski, H. Bilinski, N. Vdović, Y. Balagurunathan, E.R. Dougherty (2003) Application of image‑based granulometry to siliceous and calcareous estuarine and marine
sediments. Estuarine
Coastal and Shelf Science 58(2):
227-239.
3. H. Bilinski, S. Frančišković-Bilinski, Y. Balagurunathan, E.R.Dougherty (2003) Determination of Wadell's sphericity of sediment grains in comparison with granulometric circularity measure.
22nd IAS Meeting of Sedimentology - Opatija 2003 Abstracts book, (I. Vlahović, ed.), Zagreb, p.15.
Side Chain Control of Folding into Mixed
Peptide Helices
Carsten Baldauf, Robert Günther, and Hans-Jörg Hofmann
Institute of Biochemistry, Faculty of Life Sciences, Pharmacy and
Psychology, University of Leipzig, Leipzig, Germany
In their studies on the secondary structure formation of b-peptides, Seebach and co-workers found a novel helix type, which they named a ‘mixed helix’.1 Contrary to the
common helices, the periodicity in these helices is not realized via the monomeric constituents of the sequence, but in dimer units. This leads to an alternating formation of
hydrogen bonds between the amino acids i and (i+3) in backward (iß(i+3)) and i and (i+1) in forward (ià(i+1)) direction of the sequence and to alternating hydrogen-bonded
pseudocycles of different size (Fig. 1). In a theoretical study, we were able to show that this folding pattern can be extended to the homologous a‑, g-, and d-peptides. Even
mixed helices with still larger alternating pseudocycles with (iß(i+5)) and (ià(i+1))
interaction were identified.2
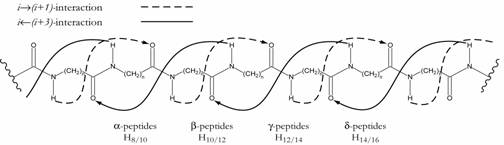
Figure 1. Hydrogen bonding pattern in mixed helices of homologous a- (n=1), b- (n=2), g‑ (n=3) and d-peptides (n=4).
Employing quantum chemical methods, we demonstrate that the stability and handedness of the different types of mixed helices can essentially be controlled by substituents at
the various backbone atoms dependent on the position and the configuration. In the case of b‑peptides, the mixed helix of the Seebach type1 is the most stable one with
alternating periodicity and even more stable than the common periodic alternatives for all substitution patterns. In g‑peptides, the common periodic structures are energetically
preferred
to the mixed representatives. However, mixed helices are generally
disadvantaged in polar solvents due to their low dipole moment.
1. D. Seebach, K. Gademann, J. V. Schreiber, J. L.
Matthews, T. Hintermann (1997) Helv. Chim. Acta 80: 2033-2038.
2.
C. Baldauf, R. Günther, H.-J. Hofmann (2004) Angew. Chem. Int. Ed. 43: 1594-1597.
PM_Match - A new way to
align RNA
structures
Stephan H. F. Bernhart1, Ivo L. Hofacker1, and Peter F. Stadler2
1Institute for Theoretical Chemistry and Structural Biology, University of Vienna, Währingerstraße 17, A-109 Wien, Austria
2Department of Computer Sciences, University of Leipzig, Kreuzstraße 7b, D-04103 Leipzig, Germany
Many classes of functional RNA molecules are characterized by highly conserved secondary structures but little detectable sequence similarity. Reliable multiple alignments
can therefore be constructed only when the shared structural features are taken into account. Since multiple alignments are used as input for many subsequent methods of
data analysis, structure based alignments are an indispensable necessity in RNA bioinformatics.
We present a method to compute pairwise and progressive multiple alignments from the direct comparison of basepairing probability matrices. Instead of attempting to
solve the folding and the alignment problem simultaneously as in the classical Sankoff algorithm we use McCaskill's approach to compute base pairing probability matrices
which effectively incorporate the information on the energetics of each sequence. A novel, simplified variant of Sankoff's algorithms can then be employed to extract the
maximum weight common secondary structure and an associated
alignment.
Solving the Multiple Genome
Rearrangement Problem with Common Intervals
Matthias Bernt, Daniel Merkle, and Martin Middendorf
Department of Computer Science, University of Leipzig, Augustusplatz
10-11, D-04109, Leipzig, Germany
The multiple genome rearrangement problem is to find a phylogenetic tree that shows how a set of genomes (sequences) may have evolved from a common ancestor. The
problem has been studied with respect to different distance measures like the breakpoint distance or the reversal distance of sequences. Biologically it is verified, that
interacting proteins are often coded by genes which are placed close to each other in the genome. This has been modelled by common intervals, which are sets of characters
(genes) that appear consecutively but possibly in a different order in all the sequences. We present a method for solving the multiple genome rearrangement problem based
on common intervals. Besides its
biological accuracy another benefit is that the computational effort for the
reconstruction can be reduced significantly.
Systematic Search of Possible Compact Nucleosome
Structures
Neva Bešker1,2, Claudio Anselmi1, and Pasquale De Santis1
1Dipartimento di Chimica, Università 'La
Sapienza', P.le A. Moro 5, I-00185 Rome, Italy
2CERM, Università degli studi di Firenze, Via
Sacconi 6, 50019 Sesto Fiorentino, Italy
Eukaryotic cells contain from 10 to 104 millions base pair in a nucleus of a few micrometers in diameter. If all the DNA molecules, which constitute a typical eukaryotic
genome would be stirred, they would span about 3 meters in length. It is therefore necessary an accurate organization of the DNA inside the cellular nuclei. Packing is due to
proteins, which fold DNA, at different levels of organization, into a structure called chromatin. The first level is well characterized: it is constituted by the nucleosome core
particles, connected together by linker DNAs. Experimental evidence suggests the structural organisation of the telomeric nucleosomes is different from the one in bulk
chromatin. We have applied the Woodcock’s two‑angles model both to telomeric and bulk chromatin. The global chromatin structure is described in terms of the rotation
angle between consecutive nucleosomes, a function of the linker length and the linker entry-exit angle. To distinguish between all the possible chromatin conformations, we
used a low resolution molecular model, based on the Gay‑Berne extension of the overlap potential for oblate ellipsoids. In the telomeric chromatin, structures with favourable
energy were found, reducing the possible conformations to two different three‑dimensional folds. In the bulk chromatin, both H1 and histone‑tails have been suggested to
bridge entering and exiting DNAs together into a stem. Histone‑tails interactions influence DNA distance, contact and parallelism and, consequently, strongly reduce the
possible chromatin conformations.
Comparative
methods for characterization of alumino‑silicates in stream sediments
Halka Bilinski1, Stanislav Frančišković-Bilinski1, Darko Hanžel2, Gábor Szalontai3, and László Horvath1
1Ruđer Bošković Institute,
HR-10002 Zagreb, POB 180, Croatia
2Institut Jožef Stefan, Jamova 39, SI-1001
Ljubljana, Slovenia
3Deptment of Silicat Chemistry and Materials
Eng., Faculty of Engineering, University of Veszprem, Veszprem, Hungary
Stream sediments are formed by weathering of rocks, which are made up of complex mixture of minerals. As major mineral products of weathering, which are not so easily
distinguished, are phyllosilicates. Their identification can give information about type of environment in which weathering occurred, namely was it acid or alkaline.
In the present work we show results obtained on several stream sediments from Croatia, Slovenia and Bosnia and Herzegovina, and also on some modeled clays. The most
powerful methods used were X-ray diffraction, Mössbauer spectroscopy and 27Al NMR techniques. The advantage of using these methods is that they do not depend on
the sediment solubility. Such multidisciplinary investigation can be used in future sediment analysis, which will be necessary due to Decision No. 2455/2001/EC of the
European Parliament and of the Council. It could also have application in planetary research, as complementary to proposed study on ocean sediments, simulating
weathering conditions on Mars.
Non-vibrational
features in NIR FT-Raman spectra of lanthanide sesquioxides
Tomislav Biljan, Zlatko Meić, and Sanda Rončević
Department of Chemistry, Faculty of Science, University of Zagreb,
Strossmayerov trg 14, HR-10000 Zagreb, Croatia
Lanthanide sesquioxides are very important compounds from technological and scientific viewpoint because of their use as starting materials in many applications of
lanthanides.1 Apparently, there have been no studies of all lanthanide sesquioxides by FT-Raman spectroscopy, except for the study of neodymium oxide.2 The purpose of
this work is to report FT-Raman and FT-NIR spectra of all lanthanide sesquioxides and yttrium sesquioxide obtained by excitation in NIR (Nd:YAG laser, 1064 nm).
The majority of lanthanide sesquioxides and yttrium sesquioxide shows additional bands in FT-Raman spectra (after the excitation with the 1064 nm line of a Nd:YAG laser)
that cannot be explained by vibrational origin. Additional bands in the FT‑Raman spectra of heavy lanthanide sesquioxides appear in the Stokes region of the spectrum, but
there are also some very strong unexpected bands in the anti-Stokes region of some light lanthanide sesquioxides and in yttrium sesquioxide, notably around 800 and
1100 cm-1. The non‑vibrational bands present in FT-Raman spectra of lanthanide sesquioxides and yttrium sesquioxide are not seen in published Raman spectra with an
excitation in visible. A possible origin of these additional bands is in luminescence of lanthanide ions.
It is important to note that interpretation of FT-Raman spectra of lanthanide sesquioxides and other lanthanide compounds should be done with great care because that
many bands are not vibrational in origin.
1.
G. Adachi, N. Imanaka (1998) Chem. Rev.
98: 1479.
2.
Y. Xu, J.Wu, W. Sun, D. Tao, L. Yang, Z. Song, S. Weng, Z. Xu, R.D. Soloway, D.
Xu, G. Xu (2002)
Chem. Eur. J. 23: 5323.
Drawing
graphs on torus
Marko Boben, Alen Orbanić, and Tomaž Pisanski
TCS, IMFM, University of Ljubljana, Jadranska 19, SI-1000, Ljubljana,
Slovenia
We present a visualization method for drawing graphs embedded in torus. The torus itself is then placed in 3-space and the whole map is represented as a 3d-graphic
object. Examples will be shown that include some well-known toroidal graphs. We will also discuss the problem of representing graph on other surfaces and give some
particular solutions that are actually used as templates for mathematical 3d-models.
The authors would like to thank Darko
Veljan for suggestion to consider knotted torus.
MixeR package for compositional data analysis
Matevž Bren1,2 and Vladimir BatagelJ2,3
1Faculty of Organizational Sciences, University of Maribor, Kidričeva 55a, Kranj, Slovenia
2Institute of Mathematics, Physics and
Mechanics, University of Ljubljana, Ljubljana, Slovenia
3Faculty of Mathematics and Physics,
University of Ljubljana, Ljubljana, Slovenia
R (http://www.r-project.org/) is `GNU S' - a language and environment for statistical computing and graphics. R is similar to the award-winning S system,
which was developed at Bell Laboratories by John Chambers et al. It provides a wide variety of statistical and graphical techniques (linear and nonlinear modelling, statistical
tests, time series analysis, classification, clustering, ...). Further extensions can be provided as packages. We started to develop the R package MixeR for the compositional
data analysis that provides support for:· operations on compositions: perturbation and power multiplication, subcomposition with or without residuals, centering of the data, solving perurbation equations,
computing Aitchison's, Euclidean, Bhattacharyya distances, compositional Kullback-Leibler divergence...· graphical presentation of compositions in ternary diagrams and tetrahedrons with additional features: barycenter, geometric mean of the data set, the percentiles lines,
marking and coloring of subsets of the data set, theirs geometric means, notation of individual data in the set ...We'll illustrate the use of MixeR with some real data.
Optimization of Empirical Force Field Parameters
for Netropsin
Urban Bren1, Jože Koller1, and Milan Hodošček2
1Faculty of Chemistry and Chemical Technology, University of Ljubljana, Aškrčeva 5, SI‑1000 Ljubljana, Slovenia
2National Institute of Chemistry, Hajdrihova
19, SI‑1000 Ljubljana, Slovenia
A netropsin molecule preferentially binds to the AT-rich domains in the minor groove of DNA and is therefore of great importance for the studies of the regulation of gene
expression. Many DNA binding ligands are also used as chemotherapeutics in pharmaceutics. Although there has been much theoretical and experimental work done on the
netropsin, we have been to our knowledge the first to optimize its empirical force field parameters. The same form of parameters for nucleic acids already exists in the
CHARMM topology and parameter files. As a result of our work we will be able to explain the binding of netropsin to the minor groove of DNA on a molecular level by
means of computer simulation techniques.
We determined partial atomic charges for the molecule using Merz-Kollman charges and the RESP model. We obtained suitable VDW parameters from the CHARMM
topology and parameter files. Bond, angle, and dihedral parameters were determined in the time consuming parameter optimization procedure by means of geometrical and
vibrational analysis of model compounds.
The results are very satisfying. The RMS deviation between netropsin geometries, optimized via ab initio and empirical methods, is only 0.58 Å. The frequencies of the
corresponding normal modes, obtained via ab initio and empirical methods, possess an average relative error of only 3.9 %. For the energy differences between the three
characteristic netropsin conformations, determined via ab initio and empirical methods, the error is lower than 0.8 kcal mole-1.
In the near future we intend to use empirical methods to calculate a vibrational spectrum of a known crystallinic form of netropsin and compare it with an experimental one.
We also want to explore netropsin's behaviour in the minor groove of DNA with the help of molecular dynamics.
Performance of Kier-Hall e-states descriptors
in QSAR of
multi-functional molecules
Darko Butina
ChemoMine Consultancy, 201 Icknield Way, Letchworth Garden City, Herts
SG6 4TT, UK
Kier-Hall e-states descriptors have been used over last 10 years in areas like predicting NMR shifts and Quantitative Structure Activity Relationships (QSAR) by the authors
of those 2D based descriptors and many other research groups. While e-states descriptors are 2D based, they have been designed to reflect electrostatic environment for
any given atom in the molecule and as such have been described as "an information rich" descriptors and therefore should have an advantage over 2D descriptors that are
based on a simple atomic/functionality type counts. The original algorithm is based on definition of 35 atomic types relevant to drug like molecules, where each atom type
has predefined intrinsic value and for each atom in the molecule, those vales are than projected onto the atom that one needs to use as a descriptor. While this approach
will work well for the problems where a single atom type is responsible for a given response, like for example, NMR shifts for series of primary amines, the problem starts
when dealing with multi-functional molecules where a single pre-defined atom type that is present more than once and in a very different environment. For example, sNH2 is
one of their predefined atom types, which represents NH2 connected to any other atom by a single bond. In a molecule that has, say three different sNH2 functionalities, like
CONH2, SO2NH2 and CH2NH2, one would than calculate an average value for sNH2 descriptor, based on three different e-states values for each sNH2. Since the same
or very similar average value for any given predefined atom type could be the result of very different chemical environment giving the same result, it will be almost impossible
for any statistical approach to resolve that problem and therefore those information rich descriptors should not outperform simple atomic counts. The performance of
Kier-Hall e-states will be tested on variety
of sets, like log P, aqueous solubility, blood brain barrier and human
intestinal absorption.
Use of chemical similarity in drug discovery
Darko Butina
ChemoMine Consultancy, 201 Icknield Way, Letchworth Garden City, Herts
SG6 4TT, UK
Chemical similarity between two or more molecules can be calculated using some sort of fingerprints and one of the many similarity indices. This talk will cover use of
Daylight fingerprints and Tanimoto similarity index. Ability of Daylight fingerprints to capture structural features of any type of molecule is unique among various molecular
descriptors that are commercially available; they do need to be ‘calibrated’ to gain the maximum information that is present in molecules of different sizes. The two most
important parameters that need to be correctly set are size of the fingerprints and the maximum lengths between atom pairs. Application of similarity will be exemplified with
clustering algorithm, dbclus, finding close analogues in HTS and from lead molecules, design of training and test sets for QSAR, and potential use in quality control, QC, of
experimental data.
Density Matrix Functional Theory
Jerzy Cioslowski1, 2 and Katarzyna Pernal2
1Department of Chemistry, Florida State University, Tallahassee, FL 32306, USA
2University of Szczecin, Department of
Physics, ul. Wielkopolska 15, 70-451 Szczecin, Poland
Density matrix functional theory (DMFT), in which the one-electron density matrix (the 1‑matrix) rather than the electron density plays the role of the main variable, holds
a promise of becoming a viable alternative to conventional electronic structure methods. The basic assumptions of DMFT are reviewed with a particular emphasis being put
on the properties of 1‑matrix functionals for the
electron-electron repulsion energy.
Three different classes of approximate functionals are discussed. The failures of the first‑generation "primitive" expressions for the electron‑electron repulsion energy are
shown and their origins are explained. The connection of the JK‑only "second-generation" functionals with the pair-excitation configuration interaction (PECI) approach is
elucidated. Other routes to
approximate implementations of DMFT are presented as well.
A Generic Framework for Geometrically Matching
Molecular Shapes
Michael Clausen1 and Axel Mosig2 1Department of Computer Science III,
University of Bonn, Bonn, Germany
2Bioinformatics, Department of Computer
Science, University of Leipzig, Leipzig, Germany
Motivated by problem settings such as the determination of motifs in proteins or molecular docking, we present a generic framework for finding geometric similarities between
two molecular shapes. Our approach is based on minimizing a distance between the two given shapes, where a problem-specific distance function can be chosen from a certain
class of
distance measures, the so-called relational
distance measures.
The setting we investigate is as follows: we are given two molecules, modeled as point sets (or, in some cases, as point sequences) P and Q in Euclidean three space, where
each point represents a chemical entity such as a single atom or an amino acid of a protein. Furthermore, we are given a distance measure d between point sets such that
d(P,Q) measures the resemblance of two molecules in a fixed spatial position, with values of d(P,Q) close to zero indicating large resemblance; the resemblance usually
changes when one of the molecules, say Q, is rotated or translated (i.e., transformed by a rigid motion g). In this setting, many typical pattern matching problems involving
molecular structures fit into one of the two following problem
settings:
Global resemblance between P and Q: It is our goal to find a transformation g that minimizes the distance between P and Q, i.e., arg ming d(P,gQ), with g ranging
over
the set RM(3) of all rigid motions in three dimensions and gQ denoting Q transformed
by a rigid motion g.
Largest common substructures of P and Q: Given a non-negative fault tolerance e, we want to determine largest possible substructures P' of P and Q' of Q such that
d(P',gQ') is at most e for some
transformation g.
In typical application scenarios, we also have physical or chemical features attached to the points in P and Q that can be taken into account by the distance measure d. Based on
algebraic considerations, our algorithms for determining global resemblance and common substructures work for any distance measure that is a relational distance measure,
and yield solutions that are within a guaranteed quality of approximation. Relational distance measures encompass distance measures such as a discrete version of the Fréchet
distance, which is a suggestive distance measure for geometrically aligning protein backbones and determining motifs, as well as the Hausdorff and the bottleneck distance,
which are well-suited for applications involving
surface interactions between molecules.
First-principles rovibrational spectroscopy
Attila G. Császár, Viktor Szalay and Gábor Czakó
Department of Theoretical Chemistry, Eötvös University, H-1117 Budapest, Pázmány sétány 1/A, Hungary
New and efficient algorithms are presented for the nearly exact solution of the few-body nuclear motion problem. These approaches, when coupled with the use of
state-of-the-art ab initio potential energy hypersurfaces, allow computation of nearly all vibrational states for small polyatomic systems, including H2O and H3+, with
exceptional accuracy.
Frequency analysis of long term
Tomislav Cvitaš1,
Nenad Kezele1,
Leo Klasinc1, Matevž Pompe2, Glenda Šorgo1, and Marjan Veber2
1Ruđer Bošković Institute, HR-10002 Zagreb, POB 180, Croatia
2Faculty of Chemistry and Chemical
Technology, University of Ljubljana, Aškerčeva 5, SI‑1000 Ljubljana,
Slovenia
Data obtained during long-term continuous ozone monitoring at different European sites were analyzed by applying Fourier transformation (FT). As expected, strong
frequency signals are found for the 1 year and 1 day periods. A collective FT spectrum for different sites was calculated. This comparison confirms the existence of a
common variation in ozone volume fractions with quasi periods ranging between 7 and 44 days. These frequencies are most probably connected with quasi-cyclic synoptic
scale meteorological influences. As expected, strong frequency signals are found for the 1 year and 1 day periods. The relative intensity of the 1 day peak can be correlated
with the intensity of local photochemical pollution as represented by a photochemical pollution index. The relative importance of the remaining peaks are calculated for each
FT spectrum and are found to describe approximately 40-44 day, 13-15 day, 8-11 day and 7 day quasi-cyclic behavior. Meteorological parameters such as temperature,
pressure and relative humidity
show some, but not perfect agreement with ozone concentration behavior.
HOW DANGEROUS IS SURFACIAL OZONE?
Tomislav Cvitaš1, Leo Klasinc1, Nenad Kezele1, Sean P. McGlynn2, and William A. Pryor2
1Ruđer Bošković Institute,
HR-10002Zagreb, POB, 180, Croatia
2
Louisiana State University, Baton
Rouge, LA 70803, USA
One hundred years after the discovery of ozone, atmospheric chemists became aware that human activities (mainly production of nitrogen oxides) have caused the
concentrations of ozone in surfacial air to increase, and that this trend continues. This increase engenders much concern because, in the entire history of Earth, life has never
experienced such elevated ozone concentrations for such long periods. Recent communications1,2 concerned with the high mortality related to air pollution in the UK and the
Netherlands during the 2003 heat wave have linked it to particulate matter and ozone concentrations. These findings raise the specter that high oxidant levels may well have
an important impact on life. Recent papers on possible ozone “production” and functions within the human body engender such suspicions3.
Based on ozone data during the heat wave 2003 as well as on monthly values of particulate matter (PM10 and PM2.5) data and mortality data for 1999-2003 in Zagreb we
can exclude ozone but not particulate matter air pollution as culprit for an excess number of deaths. Also, based on our previous results on heterogeneous lipid ozonation
with polluted air4 we present an explanation for the observation of products which seem to carry the signature of a reaction with endogeneous ozone.
1. Fischer P.H., Brunekreef B., Lebret E.
(2004) Atm. Environ. 38: 1083.
2. Stedman J.R., (2004) Atm. Environ. 38: 1087.
3. Wentworth P., Nieva J., Takeuchi C., Galve R.,.Wentworth
A.D, Dilley R.B., DeLaria G.A., Saven A., Babior B.M., Janda K.D., Eschenmoser A.,
Lerner R.A.(2003) Science 302: 1053.
4. Friedman M., Kazazic S., Kezele N., Klasinc L.,
McGlynn R.A., Pecur S., Pryor W.A. (2000) Croat.
Chem. Acta 73: 1141.
Calculation of the Optical Rotatory Dispersion of solvated molecules
by means of the
Perturbed Matrix Method
Marco D'Abramo, Alfredo Di Nola, Massimiliano Aschi, and Andrea Amadei
Università degli studi di Roma "La Sapienza", Roma, Italy
The recently introduced Perturbed Matrix Method (PMM) prooved to be very efficient in the theoretical study of electronic properties in complex molecular systems. Its
application to solvated molecules or chromophores inside a protein showed that this approach, based on molecular simulation, is relaiable at relatively low computational cost.In this work we extend PMM in order to calculate the optical rotatory dispersion of solvated molecules, using as a test system the (zwitterionic) alanine in water. Results
show that PMM procedure can provide ORD spectrum with reasonable accuracy if compared with experimental one, implying its possible use for understanding and
interpreting ORD experimental spectra of complex solute molecules, e.g. peptides and proteins.
Simultaneous optimization of exactly N‑representable density matrix
and its geminal basis
Gergely Dezső1,4, Imre Bálint1,2,3, and Iván Gyémánt1
1Department of Theoretical Physics, University of Szeged, H-6720 Hungary
2Department of Pharmaceutical Analysis, University of Szeged, H-6720 Hungary
3Department of Natural Sciences, Dunaújváros Polytechnic, H-2400 Hungary
4Department of Technology and Production Engineering, College of Nyíregyháza, H‑4401 Hungary
The most compact and complete description of quantum mechanical systems involving at most two‑particle interactions is given by the two-particle density matrices. The
two‑particle density matrix has substantially less parameters than the wave function in the same finite basis. In pursuance of quantum chemical calculations it was clarified in
the 1950s, that the density matrix can not be optimized directly because of the so‑called N‑representability problem. The N‑representability conditions needed to ensure the
exact N‑representability in the optimization procedure are unknown up to this day.A scheme for parametric optimization of the two‑electron density matrix and sample calculations are to be presented. The basic algorythm1 is surprisingly efficient in point of
the number of iteration steps.2‑4 The basic scheme is extended in a specific way. Small number of one‑electron functions are optimized in a relatively large space. The
density matrix is expanded in the basis of geminals constructed over optimized one‑electron functions. The efficiency of the method is substantially improved without
considerable increase of the number of variational parameters. The density matrix is exactly N‑representable by construction, and the procedure preserves its
N‑representability. As application, two series of correlation energy calculations were performed. The most interesting result is, that the percentage of the correlation energy
given shows saturation‑like dependence as a function of the number of optimized one‑electron function within a fixed (and larger) one‑electron space. The method is size
consistent, and full‑CI equivalent.
1. I. Bálint, G. Dezső, I. Gyémánt (2000) J. Mol. Struct.,THEOCHEM 501-502: 125-132.
2. I. Bálint, G. Dezső, I. Gyémánt (2001) J. Chem. Inf. Comp. Sci. 41: 806-810.
3. G. Dezső, I. Bálint, I. Gyémánt (2001) J. Mol. Struct. THEOCHEM 542: 21-23.4. I. Bálint, G. Dezső, I. Gyémánt (2001) Int. J. Quant. Chem. 84(1): 32-38.
Perfect Clar
Structures by Capra Operation
Mircea V. Diudea1, István Lukovits2, and Ante Graovac3,4
1Faculty of Chemistry and Chemical Engineering,
“Babeş-Bolyai” University,
RO-400084 Cluj, Romania
2Central
Research Institute for Chemistry, Hungarian Academy of Sciences, H-1525
Budapest, P.O.Box 17, Hungary
3Ruđer Bošković Institute, POB 180,
HR-10002 Zagreb, Croatia
4Faculty of Natural Sciences, Mathematics and
Education, N. Tesle 12, HR-21000 Split, Croatia
The third basic operation on maps obeying the Goldberg1
multiplication rule: ![]() , predicts our “Capra”
Ca-operation2 for the series: Le: (1, 1),
, predicts our “Capra”
Ca-operation2 for the series: Le: (1, 1),
m = 3; Q: (2, 0), m = 4 and Ca: (2, 1), m = 7. Ca-operation insulates each parent face
by its own hexagons (i.e.,
corannulene substructures), in contrast to Le
and Q.
In the present paper it is shown that Ca-operation produce, when applied on Clar polyhedra (i.e., those having perfect Clar structures, PCS), objects having a disjoint set of
corannulenoid regions (Figure 1).
C420, PCS C420, PCorS


Any perfect Clar-like corannulenoid structure PCorS is a PCS. The Capra transform of a convex polyhedron or a polyhex torus is a PCorS if and only if it is a PCS. Such
structures can be named
fully-resonant-corannulenoid molecules,
and there are expected to be maximally stable in a localized valence bond
picture.3
1. M. Goldberg (1937) Tôhoku Math. J. 43:
104-108.
2. M.V. Diudea (2003) Studia Univ. “Babes-Bolyai”
48 (2): 3-16.
3. J.R. Dias (1987), Thermochim. Acta 122:
313-337.
Protein structure modeling and the Protein
Structure Initiative
András Fiser
Department of Biochemistry & Seaver Center for Bioinformatics, Albert
Einstein College of Medicine, 1300 Morris Park Ave, Bronx, NY 10461, USA
Functional characterization of proteins is one of the most frequent problems in biology. While sequences provide valuable information, their high plasticity makes it frequently
impossible to identify functionally relevant residues. Functional characterization of a protein is often facilitated by its three dimensional (3D) structure. Genome scale
sequencing projects have already produced more than a 1.5 million unique sequences, while only 25000 of these have their 3D structures solved experimentally using X-ray
crystallography or NMR spectroscopy. Because of the inherently time consuming and complicated nature of structure determination techniques, the fraction of experimentally
solved 3D models is expected to further shrink from the current level of less than 2%. Computational approaches need to be employed to bridge the gap between the
number of known sequences and that of 3D
models.
We briefly overview the current approaches to protein structure modeling with an emphasis on methodological details of comparative modeling techniques. Next, our
ab initio loop modeling method will be discussed along the lines of refining comparative protein models. Finally, the use of structure modeling methods will be discussed
through specific biological applications and in the
context of the world wide "Protein Structure Initiative".
A mathematical model relating stream sediment geochemistry and the Kupa River
drainage basin lithology
Stanislav Frančišković-Bilinski
Ruđer Bošković Institute, POB 180, HR-10002 Zagreb, Croatia
This study (from planned dissertation of author) constitutes the first geochemical investigation of stream sediments in the Kupa River drainage basin, which area covers
10052 km2 and is situated in three countries (Croatia, Slovenia, Bosnia and Herzegovina). In order to assure the same sedimentary conditions, sediments have been sampled during the dry season in June - August 2003. Major- (8) and trace- (45) elements were
determined by ICP-MS method (in Actlabs, Canada) in fraction <63 ľm at 63 locations. Bedrock lithology (44 units), which represents the parent material for the
sediments, was determined for each of the 63 drainage subcells, using Geological map 1:500000, sheet Zagreb. The drainage basin was delineated in such a way that each
basin segment corresponded to the area upstream and upslope of each sampling site. As a result of vectorization, topology and calculated surfaces, percentage of lithological
units were obtained for each subcell. Basic statistics (program Statistica 6.0) was used to find marked positive correlations of major and trace elements with corresponding
lithologies. From 44 lithological units, 8 reduced lithologic variables were specified and used in factor analysis. The geochemical variables included a selection of 8 major and
17 minor and trace elements, similarly as performed by Halamić et al. (2001) for Medvednica Mt.To illustrate how computers could help the modern geologists and how difficult it is to get exact information from a multivariate method, such as factor analysis, 4 calculation
experiments have been performed. Lithologic variables were specified in different ways:
1. according to age of formation
2. according to related rocks, independent on age
3. according to related rocks, but taking into account correlation of elements and all 44 lithological units
4. according to computer based clustering of lithological units.
Results will be presented for all 4 cases, showing relationships of elements and lithologic units, reduced into several geologically meaningful factors. The differences presented
for 4 cases will be discussed and the best model suggested.
J. Halamić, Z. Peh, D. Bukovec, S. Miko, L. Galović (2001) Geologia Croatica 54/1: 37-51.
Distribution of SNPs in
Genes, Phylogenetic Footprints and Non-Coding Regions
Claudia Fried1,2, Peter Ahnert3, and Peter F. Stadler1,2
1Institute of Informatics, University of Leipzig, Kreuzstraße 7b, D-04103 Leipzig, Germany
2Institute of
Theoretical Chemistry and Structural Biology, University of Vienna, A-1090,
Vienna, Austria
3IKIT/BBZ, Faculty of Medicine, Faculty of Medicine, University of
Leipzig, D-04103 Leipzig, Germany
Recently it has been shown that different patterns of polymorphisms between the coding and non‑coding regions seem to distinguish functionally different gene groups e.g.,
immune genes dealing with self molecules, and immune genes targeted towards foreign molecules. It is likely that systematic patterns in the distribution of polymorphism
between coding, non-coding but regulatory functional, and non-functional sequences can be found also in other classes of protein-coding genes. Here we present a systematic
computational approach to address this problem.DNA sequences that are regulatory active presumably cover a substantial part of intergenic regions. On the other hand, only a
small number of transcription factor binding sites or promoter sequences are experimentally verified for any given gene. Fnctional non-coding sequences evolve much slower
than the surrounding non-functional DNA because they are subject to stabilizing selection. Comparative sequence analysis can therefore be used to detect functional non-coding
DNA sequences in the vicinity of the genes of interest. This technique is known as phylogenetic footprinting. Recently we have presented the program tracker as an efficient
tool for surveying phylogenetic footprints in large datasets. For the purpose of the present study we used tracker to detect phylogenetic footprints in human genes with homologs
in mouse, zebrafish and pufferfish. In all cases the DNA sequence extending 10000nt upstream and downstream of the gene was retrieved via ENSMART.
SNP density was calculated for genes (including, exons, introns and untranslated regions), phylogenetic footprints and for non-coding sequences. The SNP density was
compared between genes and non-coding sequences and between phylogenetic footprints and non-coding sequences. Fishers exact test was used to determine the significance
of differences.
Intuitively one would expect that the rate of occurrence of SNPs in the non-functional DNA is largest since there it is no subject to selection, while mutations should be selected
against in coding and regulatory
sequences. On average for all 8213 human genes considered here, SNPs indeed appear to be significantly underrepresented in genes and phylogentic footprints opposed to
non‑coding sequences. However, some genes show a overrepresentation of SNPs in phylogenetic footprints or higher underrepresentation of SNPs in phylogenetic footprints
than other genes. To answer the question whether the density of SNPs in genes, phylogenetic footprints, and non‑functional background provides information on selection
pressures acting on various group of genes we will assign the genes to their Gene Ontology terms.
Quantitative individual based models of
multicellular growth processes?
Jörg Galle1 and Gernot Schaller2
1Interdisciplinary Centre for Bioinformatics, University Leipzig, Leipzig, Germany
2Institute of Theoretical Physics, Technical
University, Dresden, Germany
A variety of physical and biological models have been developed in the past to examine the principles of the spatiotemporal organisation of cell populations. Due to the
recent advances in biophysics and cell-biology the possibilities to collect new information about specific parameters on cells and tissues are strongly improving. In
conjunction with these possibilities new perspectives arise to introduce quantitative models of the multicellular growth processes. Individual based off-lattice models
represent a novel trend in this context. However, the question remains whether or not these models do actually allow extending the scope of computer studies to predict cell
population behaviour?
We will compare two novel approaches recently introduced by us; modelling cells either by deformable spheres or as deformable Voronoi polyhedra.
PROTEIN TRANSDUCTION AS A TOOL FOR
DELIVERING OF DRUGS INTO THE CELLS
Mira Grdiša
Division of Molecular Medicine, Ruđer
Bošković Institute, POB 180, HR-10002 Zagreb, Croatia
Delivery of macromolecular drugs (e.g. antisense oligonucleotides, polymer-drug conjugates, etc.) designed to work in specific sites inside cells is complicated as
macromolecules typically have access to fewer biological compartments than small molecules.To achieve an efficient intracellular drug delivery into the cells, TAT peptides derived from the HIV-1 TAT protein facilitate intracellular delivery of proteins and small
colloidal particles. TAT fusion protein enters into the cells when added to the surrounding media, in concentration depended manner. A TAT component is degraded by
proteolytic enzymes, protein is refolded with cell chaperons (Hsp proteins) and ready for physiological function.In general, plasma membranes of cells are impermeable to proteins and peptides. Because of that, the potential for intracellular therapeutic use of proteins, peptides and
oligonucleotides has been limited by the impermeable nature of the cell membrane to these compounds. Thus protein transduction has been overcame that obstacle, and it
has been widely used to analyze biochemical processes in living cells. The present study showed how transduced proteins influence on the regulation of cell cycle. The
proteins (p27, p23, Mp27) were transdused into different cell lines (NALM, MOLT, Raji, SuDHL, and K562) and their effects on proliferation of the cells were measured.
A transduced p27 did not remarkable influence on proliferation of examined cell lines. Mutated p27 inhibited the proliferation of examined cell lines up to 30 %. On the other
hand, a transduction of p23 protein, truncated form of p27, inhibited the proliferation all of examined cell lines 30-60 %. Also the effects on expression of host p27 protein
were examined, as well as an influence on induction of apoptosis.
Connecting Biology & Chemistry - Using
Ontologies
Jürgen Harter
BioWisdom Ltd., Babraham Hall, Babraham, CB2 4AT Cambridge, U. K.
People use the word "ontology" to mean different things, e.g. glossaries & data dictionaries, thesauri & taxonomies, schemas & data models, or formal ontologies &
inference. This presentation explains what an ontology is and how it can be used within the life sciences domain in particular pharmaceutical R&D. It concentrates on
showing the benefits of employing ontologies for integrating chemistry and biology data. An upper ontology for the chemistry and biology domain will be shown as an
example. A knowledge map for chemistry will illustrate the links between compounds, drugs, pharmacology, disorders, side effects, toxicology, molecular properties etc.
Some use cases for ontologies will be discussed, in particular the sort of questions that can now be answered by having an ontology: e.g. which brain-specific proteins are
the targets for established marketed drugs? Furthermore, various cheminformatics and bioinformatics software tools that can interact with an ontology knowledge server will
be explored, thus providing a mechanism to explore similarity searching for example, or other statistical analyses like cluster analyses.
A tutorial on RNA Secondary Structure
Prediction
Ivo Hofacker
Institute
for Theoretical Chemistry and Structural Biology, University of Vienna,
Währingerstraße 17, A-1090, Vienna, Austria
This tutorial give a practical introduction to RNA secondary structure prediction and analysis, using the tools provided by the Vienna RNA package.
We'll briefly present the energy model for RNA secondary structures and outline the dynamic programming solution to the folding problem.
We'll then present tools to solve typical structure prediction tasks, such as:
- Prediction of optimal RNA secondary structures
- Predicting structural alternatives and equilibrium ensembles
- Assessing the reliability of predicted structures
- Comparing and aligning secondary structures
- Predicting consensus structures and detecting functional structures
- Simulating folding dynamics and analysis of energy landscapes
Finally, we'll show how to build custom applications for specific problems using the Vienna RNA code library.
Database of pore domain models from cationic
channels
Ana Jerončić and Davor Juretić
Faculty of Natural Science and Mathematics, N. Tesle 12, HR-21 000
Split, Croatia
In the post-genomic proteome era, biologists often need to construct their own problem‑specific databases in order to support the analysis and knowledge discovery. In this
context, the database of channel proteins, with accurate structure and functional annotations, is of special interest in neuroscience, highly dynamic and overwhelming research
area. Here we report a customized database of pore‑forming domains from cationic channels (the P-region domains) that was created by using semi‑automated collection
and management as well as high accuracy modeling procedure. Literature and data mining problems are exposed in connection with collecting the unique set of functional
channel protein families. After clarifying these problems, the family-specific potentials for identification of new members are identified from database’s current state of
channel‑protein knowledge. In the absence of more than handful of high resolution 3D structures, sequence analysis techniques are the sole solution for pore domain
definition. Therefore, we built pore domain models, with modeling procedure that was based on use of several bioinformatics tools, developed with primary sequence input,
and evaluated with appropriate statistical techniques. Modeled data are stored in a MySQL database which can be interactively queried over the web at
URL: http://www.pmfst.hr/~alucin/PPore
Enzyme evolution and entropy production
Davor Juretić and Paško Županović
Faculty of Natural Sciences, Mathematics and Education, University of
Split, N. Tesle 12, HR‑21000 Split, Croatia
Physicists are well aware that variational principles are very powerful way of describing the nature. There is no a priori reason why variational principles should not be
applied to gain better understanding of biological evolution as well. The minimum entropy production theorem, promoted by Ilya Prigogine, has been the prime candidate for
the variational principle from physics relevant for the description of biological phenomena. Recently, Roderick Dewar has derived the maximum entropy production principle,
as the general selection principle for nonequilibrium stationary states. In this work we ask the question about present day free energy conversion performed by membrane
enzymes important in bioenergetics. When they establish the stationary state is it closer to minimum or to maximum entropy production state? We show that such enzymes
operate far from the thermodynamic equilibrium and much closer to the maximum then to the minimum entropy production state. From this result one can assume that
biological evolution gradually produced structures designed to keep the system far from equilibrium, when associated dissipation must be high. High dissipation means that
the thermodynamic evolution is accelerated in the presence of life. In other words, from the point of view of a physicist, increased entropy is identical goal for the
thermodynamic and biological evolution.
Synthesis, Spectroscopic Characterization and Biological Activity of
N-1-Sulfonylcytosine
Derivatives
Jelena Kašnar-Šamprec1, Ljubica
Glavaš-Obrovac2,
Marina Pavlak3, Nikola
Štambuk4, Paško Konjevoda4, and Biserka Žinić4
1Children’s Hospital Zagreb, Zagreb, Croatia
2Department of Nuclear Medicine and Pathophysiology ,
Clinical Hospital Osijek, Croatia
3Department of Biology, Faculty of Veterinary Medicine, Zagreb University, Zagreb, Croatia
4Ruđer Bošković Institute, Zagreb,
Croatia
N-sulfonylpyrimidine derivatives were designed and synthesized as a new type of sulfonylcycloureas.1,2 We will describe the large scale preparation of N-1-sulfonylcytosine
derivatives, which have been characterized by the spectroscopic methods. The aim of the present study was to investigate in vivo antitumor activity of novel N-1-sulfonylcytosine
derivatives 1-(p-toluenesulfonyl)cytosine (4H), 1-(p-toluenesulfonyl)cytosine hydrochloride (4HxHCl). These types of compounds showed potent inhibitory activity on the growth
of human tumor cell lines in vitro. In comparison with 5-fluorouracil some of N‑sulfonylpyrimidine derivatives showed 10 times stronger activity and some of them showed the
ability to induce apoptosis in treated tumor cells. The inhibitory effect of the investigated derivatives on normal human cell lines was lower in comparison to antitumor effects.3
In addition to the antitumor effects hematologic findings following the
parenteral administration of substances were also investigated.

1. B. Kašnar, I. Krizmanić, M. Žinić
(1997) Nucleosides & Nucleotides 16: 1067-1071.
2. B.
Žinić, M. Žinić, I. Krizmanić (2003) Sulfonylpyrimidine derivatives with anticancer activity, EP
0 877 022 B1, 16.04.2003.
3. Lj. Glavaš-Obrovac, I. Karner, B. Žinić, K.
Pavelić (2001) Anticancer Res. 21: 1979-1986.
Investigation of liquid and gas phase ligation of metal ions with crown ethers
by mass spectrometry
Saša Kazazić1,
Leo Klasinc1, Bogdan
Kralj2, Dunja Srzić1, Ljerka Tušek‑Božić1, and Dušan žigon2
1Ruđer Bošković Institute, HR-10002
Zagreb, POB 180, Croatia
2Department for Environmental Sciences, Jožef
Štefan Institute, Jamova 39, SI-1000 Ljubljana, Slovenia
Liquid phase and gas phase metallation of following crown ethers: 18-crown-6 (18C6), dibenzo-18-crown-6 (DB18C6), bis(4-tert-butylbenzo)-18-crown-6
(mtb-DB18C6), bis(3,5‑di-tert-butylbenzo)-18-crown-6 (dtb-DB18C6) and bis(4‑hexadecylbenzo)-18-crown-6 (hd-DB18C6) with Li+, Na+, K+, Rb+ and
Cs+ ions was investigated by fast atom bombardment (FAB), electrospray ionization (ESI) and laser desorption/ionization Fourier transform (LDI-FT) mass
spectrometry. Special attention was paid to the relative rates of formation and stability of monomeric products, as well as to the addition of a second crown ether
molecule
to them.
On
visualizing carbon cages
Edward
C. Kirby
Resource Use Institute, 14 Lower Oakfield, Pitlochry, Perthshire PH16
5DS, Scotland UK
The chemical graphs of carbon cages constructed from graphite‑like sheets – fullerenes, nanotubes, toroidal structures and the like – are often extraordinarily difficult to fully
comprehend if presented as simple wire-frame images in a 3D perspective. Relative movement such as rotation of the image helps a lot, and a number of software tools allow
this to be done on a computer screen. However, with the current state of technology, this is of no help towards presenting comprehensible diagrams for the printed page.
Following a few other contemporary papers, we have, in recent work on fully‑resonant‑azulenoid systems (Kirby, submitted), experimented with the selective colouring of
faces to improve clarity, and some examples of this class are shown here. Some of these objects also have a certain aesthetic appeal.
Fibonacci cubes are the resonance graphs of
fibonaccenes
Sandi Klavžar and Petra
Žigert
Department of Mathematics, PeF, University of Maribor, Koroška c. 160,
SI‑2000 Maribor, Slovenia
Fibonacci cubes were introduced in 1993 and intensively studied afterwards. Fibonacci cubes are precisely the resonance graphs of fibonaccenes. Fibonaccenes are graphs
that appear
in chemical graph theory and resonance graphs reflect the structure of their
perfect matchings. Some consequences of the main result will also be presented.
Met-enkephalin and Naloxone Effects on Head and Neck Squamous
Cell Carcinoma Cell Lines
Sandra Kraljević, Nikola Štambuk, Marijeta Kralj, and Paško Konjevoda
Ruđer Bošković Institute, Bijenička cesta 54, HR-10002
Zagreb, Croatia
Met-enkephalin is a neuropeptide with multifunctional properties on cell growth, division and differentiation. Its effects include in vitro and in vivo modulation of tumor cell
growth via delta and/or zeta opioid receptors. We investigated the effects of met-enkephalin and naloxone (opioid receptor antagonist) on cell lines CAL-27 and Detroit-562
of the head and neck squamous cell carcinoma (HNSCC). The influence of met-enkephalin and naloxone on tumor cell lines growth was evaluated using MTT cell
proliferation assay. Tested substances were applied in the range of pharmacologically relevant concentrations (10-6 to 10-3 M). Dose-response curve analysis of
met-enkephalin and naloxone effects on the tumor cell growth was done by means of GraphPad Prism Software, version 4.0. We found that opioid receptor blockade was
the
primary mechanism of agents’negative influence on CAL-27 and Detroit-562
cell line growth.
Evolution of artificial gene regulation networks for the control of
cell-motion and
cell-cell-interaction
Matthias Kruspe1 and
Dirk Drasdo2
1 University of Leipzig,
Department of Bioinformatics, Inselstraße 7b, D-04103 Leipzig, Germany
2 Max-Planck-Institute of
Mathematics in the Sciences, Inselstraße 22-26, D-04103 Leipzig, Germany
Despite the spectacular progress in biophysics, molecular biology and biochemistry our ability to predict the dynamic behavior of multicellular systems under different
conditions is very limited. One reason for this is that, different from simple physical particles, cells can change their properties due to genetic or metabolic regulation. The
rules that underlay the regulation have been determined on the time scale of evolution, typically by selection on the phenotypic level of cells or cell populations.
While most approaches attempt to understand the principles underlying intracellular control processes from the perspective of the single constituents, we in this paper adopt
the perspective of a bird's eye view.
We illustrate by in-silico simulations how networks that control cellular behavior may develop as a consequence of an artificial evolutionary processes, if either the cells, or
populations of cells are subject to selection on particular features. The networks is encoded by binary strings, which may be considered as encoding the genetic information
(the genotype) and are subject to mutations and crossovers. The cell behavior reflects the phenotype. We consider two examples, migration strategies of single cells
searching a signal source, and cell-cell aggregation.
We find that the networks that are selected during the artificial evolutionary process encode naturally found migration and aggregation strategies such as a random walk,
systematic deterministic search, chemotaxis and chemokinesis.
Topological description of single‑wall carbon
nanotube junctions
István László
Department of Theoretical Physics, Budapest University of Technology and Economics,
BUTE Center for Applied Mathematics and Computational Physics,
H-1521 Budapest, Hungary
Carbon nanotube junctions have emerged as good candidates for building blocks in nanosize networks. They are also interesting for their potential use in nanoscale transistor
or amplifier applications.Single-wall carbon nanotubes can be either metallic or semi-conducting depending on both the diameter and chirality. Heterojunctions formed by a
semi conducting and a metallic nanotube have rectifying properties.More sophisticated devices can be constructed by applying n-terminal junctions with n > 2. In this work
an
algorithm is presented for generating various junctions between nanotubes of
different chirality and diameter. We shall study Y, T, X and other junctions.
Love in van der Waals equation
M. Howard Lee
Department of Physics, University of Georgia, Athens, 30602 GA, USA
The equation of state bearing the name van der Waals is known to almost everyone in physics and chemistry. It was proposed in 1873, before the old quantum theory and
long before modern statistical mechanics. Yet it has remained a useful and popular model to this date, more than a hundred years later. The main appeal perhaps is its
simplicity in describing the gas liquid transition that is not surpassed by the modern theories. Can one say anything new about this model now? Probably not likely except
perhaps, put it figuratively, love - almost like a love triangle – contained in a cubic equation that characterizes this model. The solutions are among the loveliest in the literature
of cubic equations.
Faber-Krahn Type Inequalities for Trees
Josef Leydold and Türker Biyikoglu
Institute of Statistics, Vienna University of
Economics and Business Administration, Augasse 2-6, A-1090 Vienna, Austria
The Faber-Krahn theorem states that among all bounded domains with the same volume in Rn (with the standard Euclidean metric), a ball that has lowest first Dirichlet
eigenvalue. Recently it has been shown that a similar result holds for (semi-)regular trees. In this talk we show that such a theorem also hold for other classes of (not
necessarily regular) trees. However, for these new results no counterparts
in the world of the Laplace‑Beltrami‑operator on manifolds are
known.
Generation and symmetry classification of 2‑cell embeddings:
A method to predict new
molecular structures
Erwin Lijnen and Arnout Ceulemans
Departement Chemie, K.U. Leuven, Celestijnenlaan 200F, B-3001 Leuven,
Belgium
The study of highly symmetrical graphs offers interesting perspectives for the design of new molecular frameworks. The molecular realization of a graph requires that its
vertices are positioned in three-dimensional space. In this process topological aspects of graph theory become important.1 We are especially interested in molecular
realizations where the graph is mapped on a closed surface, like a sphere or torus. This process is called an embedding. In the present contribution we examine the set of all
possible embeddings that a given graph may have. This set may be very large and can have an interesting and rich structure. In the chemical literature it is not realized that
combinatorial techniques have been described which can retrieve all possible embeddings of a graph.2 We present an algorithm that makes us of group theory to generate all
distinct embeddings of a graph. The strength of this algorithm is that it greatly reduces the number of embeddings by removing symmetry redundancies. We illustrate our
procedure for the highly symmetrical Möbius‑Kantor graph with 16 vertices. In principle this graph has 216 embeddings but symmetry factorization reduces this number to
only 759 non-equivalent embeddings. The most symmetrical embeddings are further investigated and proposed as building blocks for new types of negatively curved carbon
allotropes. 3
Financial support from the Fund for Scientific Research –
Flanders (FWO) is gratefully acknowledged.
1. Gross J.L., Tucker T.W. (2001) Topological Graph Theory, Dover Publications Inc., New York.
2. Mohar B.,
3. Lijnen E., Ceulemans A., Oriented 2-cell embeddings
of a graph and their symmetry classification: generating algorithms and case
study of the Möbius-Kantor graph. Submitted to: J. Chem. Inf. Comp. Sci.
The differentiation of virus strains by restriction enzyme analysis and
determination of strain similarities
Ivana Lojkić1, Paško Konjevoda2, Zdenko Biđin1, and Biserka Pokrić2
1Faculty of Veterinary
Medicine, University of Zagreb, HR-10000 Zagreb, Heinzelova 44, Croatia
2Ruđer Bošković Institute, HR-10002 Zagreb, POB 180,
Croatia
The differentiation of 35 field isolates of the infectious bursal disease virus (IBDV) was carried out by the restriction enzyme (RE) analysis of the products obtained by
reverse transcription (RT)/polymerase chain reaction (PCR). The 422‑bp hypervariable region of the VP2 gene (nucleotides 732-1153), amplified by RT/PCR, was
digested using the restriction endonucleases CfoI, SacI, SspI, StuI and TaqI. The knowledge of the nucleotide sequence recognized by the endonuclease, the literature and
the GenBank nucleotide data for the analyzed VP2 gene region, as well as the size of the fragments obtained by the endonuclease digestion, enabled the determination of the
nucleotide position corresponding to a restriction site. The differentiation of the analysed IBDV species was obtained by comparing the position of the restriction sites in
analysed IBDV species
with the data reported elsewhere.
The experimentally obtained results were analysed by partition around medoids (PAM) and monothetetic clustering (MONA) algorithms for unsupervised learning. Five
groups of viral strains were
extracted by both methods and the viral strains classification based the
restriction site similarities is presented by the banner plot of PAM
method.
Improved structure-toxicity relationships for
aquatic toxicity of environmental pollutants
Bono Lučić1, Dragan Amić2, Marjana Novič3, Damir Nadramija4, and Ivan Bašic4
1Ruđer Bošković Institute, P.O. Box
180, HR-10002 Zagreb, Croatia
2The Josip Juraj Strossmayer University, P.O. Box 719, HR-31001 Osijek, Croatia
3National Institute of Chemistry, P.O.B. 660, SI-1001 Ljubljana, Slovenia
4PLIVA Hrvatska d. o. o., GBS-Life Science,
Prilaz b. Filipovića 25, HR-10000 Zagreb, Croatia
Numerous organic chemicals can be environmental pollutants, and due to this fact, many studies were directed towards the understanding of relationships between a structure
and toxicity of a compound. Structure-toxicity models are strongly dependent on the class of molecules for which models are obtained. Classification of molecules is defined
by the mechanism of the toxic action of molecules, and this piece of information can be obtained experimentally, or predicted by developed algorithms.1,2 We will test the
degree of improvement of existing models for predicting toxicity of molecules by using structural descriptors computed by the Dragon program3, and by including information
on the mechanism of toxic action.
Analysis was based on 293 organic molecules for which experimental aqueous toxicity on Poecilia reticulata were collected from the literature.4 Starting from structures
encoded as the SMILES string, molecular structures were generated by the CORINA 3D structure generator (www2.chemie.uni-erlangen.de/software/corina/). Molecules
were characterized by more than 800 molecular descriptors that were filtered in order to remove highly inter-correlated descriptors. Final models were selected by the
CROMRsel program for efficient selection of a small sub-set of the most important descriptors into the multiregression models. We obtain simple multivariate form of models
containing 2-8 optimized parameters (i.e. 1-7 of descriptors) with
better statistical performance than the published models developed on the same
set of molecules.
1. O. Ivanciuc (2002) Internet Electron. J. Mol. Des.1: 157.
2. A.O. Aptula et al. (2002) Quant. Struct.-Act. Relat. 21: 12.
3. R. Todeschini, V. Consonni: http://www.disat.unimib.it/chm/
4. A.R. Katritzky et al. (2001) J. Chem. Inf. Comput. Sci. 41: 1162.
Simplified structure-solubility relationships for early
ADME evaluation in drug discovery
Bono Lučić1, Damir Nadramija2, Ivan Bašic2, Dean Nasteski3, Dragan Amić4, and Nenad Trinajstić1
1Ruđer Bošković Institute, P.O. Box 180, HR-10002 Zagreb, Croatia
2PLIVA Hrvatska d.o.o., GBS-Life Science,
Prilaz b. Filipovića 25, HR-10000 Zagreb, Croatia
3Brune Bušića 15, HR-10000 Zagreb,
Croatia
4The Josip Juraj Strossmayer University, P.O.
Box 719, HR-31001 Osijek, Croatia
Relationship between molecular structure and solubility of 1039 organic compounds were studied. Initial structures of molecules were encoded as SMILES and converted to the
3D structures by the CORINA program (www2.chemie.uni-erlangen.de/software/corina/) and more than 1000 initial descriptors were computed by the program Dragon 2.1.1
Initial set of descriptors was filtered in order to remove non-significant and highly inter-correlated descriptors (123 descriptors remained after filtering). Finally, the best linear
multiregression models containing 1-7 descriptors were selected from the set of 123 descriptors by the CROMRsel program.2 Standard error of estimate and standard error of
leave-one-out cross-validation
obtained on the training set are 0.739 and 0.746 log units, respectively.
In the best seven-descriptor model five topological descriptors, one atom-centered fragment descriptor and calculated Moriguchi octanol-water partition coefficient are involved.
Using the best linear seven‑descriptor model we performed prediction on external data set containing 258 molecules. We obtained the same prediction of solubility for 258
molecules from the test set (Stst = 0.745 log units), as it was obtained by the neural network model developed on the same sets of molecules.3 It should be mentioned that the
statistical parameter (Stst = 0.71 log units) given in Table 1 (page 1636) for the best neural network model was not computed correctly – correct value is Stst = 0.745 log units.
However, our seven-descriptor model is linear (containing only eight optimized parameters) and much simpler than the above mentioned neural network model (which is a
nonlinear model, containing seven descriptors and having 19 optimized parameters). Presented linear multiregression models can be additionally improved by the inclusion of
nonlinear terms of initial descriptors.
1. http://www.disat.unimib.it/chm/
2. B. Lučić, N.
Trinajstić (1999) J. Chem. Inf.
Comput. Sci. 39: 121-132.
3. R. Liu, S.-S. So (2001) J. Chem. Inf. Comput. Sci. 41: 1633.
Resonance Energy in Graphite
István Lukovits
Chemical Research Center, H-1525 Budapest, POB 17, Hungary
According to Zhu et al.1 the resonance energy/electron (REPE) is, equal to 0.17 eV in rectangular graphite sheets graphite sheet. Only π-electrons were considered. In this
work REPE was calculated for parallelogram-shaped graphite sheets (PSGSs). The number of Kekulé structures and contributions of benzene‑like and naphthalene‑like
conjugated circuits were taken into account.
It was found that, REPE = 0.00 eV in PSGSs. The convergence is slow. These results indicate that PSGS is less "aromatic" than its rectangular counterpart like in the
polyacene/polyphenanthere case.
Therefore addition reactions are expected to be less difficult to carry out in
PSGSs than in graphite sheets of rectangular shape.
1.
H.-Y. Zhu, A.T. Balaban, D.J. Klein, T. Živković (1994) Conjugated-circuit
computations on two dimensional carbon networks. J. Chem. Phys. 101:
5281-5292.
Combines Phylogenies: Hosts and their
Parasites
Daniel Merkle and Martin Middendorf
Department of Computer Science, University of Leipzig, Augustusplatz
10-11, D-04109 Leipzig, Germany
Hosts and their parasites are prominent model systems for studying coevolutionary processes. A fundamental problem in the theory of comparing host-parasite phylogenies is
the reconstruction of past associations between hosts and parasites. Event based methods for solving the reconstruction problem take advantage of knowledge about the
likelihood of possible evolutionary events. In this talk we give a short overview over event based methods for the reconstruction problem and present a tool that we have
developed for solving this problem when extinction
events are possible.
Coding and Ordering Kekulé Structures
Ante Miličević, Sonja Nikolić, and Nenad Trinajstić
Ruđer Bošković Institute, HR-10002 Zagreb, POB 180, Croatia
The concept of numerical Kekulé structures is used for coding and ordering geometrical (standard) Kekulé structures of several classes of polycyclic conjugated molecules:
catacondensed, pericondensed, and fully arenoid benzenoid hydrocarbons, thioarenoids, and [N]phenylenes. It is pointed out that the numerical Kekulé structures can be
obtained for any class of polycyclic conjugated systems that possesses standard Kekulé structures. The reconstruction of standard Kekulé structures from the numerical
ones is straightforward for catacondensed systems, but this is not so for pericondensed benzenoid hydrocarbons. In this latter case, one needs to use two codes to recover
the geometrical Kekulé structures: the Wiswesser code for the benzenoid and the numerical code for its Kekulé structure. There is an additional problem with pericondensed
benzenoid
hydrocarbons; there appear numerical Kekulé structures that correspond to two
(or more) geometrical Kekulé structures!
However, this problem can also be
resolved.
Quantization of electrons in ultrathin
metallic films
Milorad Milun
Institute of Physics, Bijenicka 46, HR-10002 Zagreb, Croatia
Under specific conditions ultra-thin metallic films may serve as a potential well for electron confinement. The confinement leads to quantization of electron energy, i.e., defines
the allowed electron energy states and these may be detected and studied by photoelectron spectroscopy, inverse photoemission spectroscopy and Scanning tunneling
microscopy.
These films being only a few atoms thick are very useful example of one‑dimensional nanostructures whereas the system remains macroscopic in remaining dimensions. It has
been observed in many cases an oscillatory behaviour of magnetic and electronic properties of such nano‑structured layered metallic systems.
The spectroscopy of quantum well states provides information about the electronic properties of these nano‑structures. In some cases a huge difference in the properties of
just‑a‑few‑atoms thick and thicker, say 6 and more atoms, films was found. Such phenomena are described and discussed in this talk with an emphasis to the lowest film
thicknesses.
SYMMETRY PROPERIES OF SOME MOLECULES
Ghorban Ali Moghani1 and Ali Reza Ashrafi2
1Department of Mathematics, Faculty of Science, Payame Noor University, Tehran, Iran
2Department of Mathematics, University of
Kashan, Kashan, Iran
By symmetry we mean the automorphism group symmetry of a graph. The symmetry of a graph, also called a topological symmetry, accounts only for the bond relations
between atoms, and does not fully determine molecular geometry. The symmetry of a graph does not need to be the same as (i.e. isomorphic to) the molecular point group
symmetry. However, it does represent the maximal symmetry which the geometrical
realization of a given topological structure may posses.
In this paper, we present a fast
algorithm to compute the symmetry properties of some molecules.
Keywords: automorphism group, symmetry property, point group.
ACIDITY OF LINEAR POLYCYCLIC
TETRAZOLE COMPOUNDS
Svetlana E. Morozova, Andrei V. Komissarov, Kirill A. Esikov, and Vladimir A. Ostrovskii
Saint Petersburg State Institute of
Technology (Technical University), 26, Moskovsky prosp., Saint Petersburg,
190013, Russia
Usually polycyclic tetrazole compounds are capable to show high complexing activity in relation to ions of different metals. We obtained series of linear polycyclic tetrazole
compounds containing three tetrazole cycles, one
of which is NH-unsubstituted, and corresponding carboxylic acids.
|
N1-isomers (cycle B) |
 |
|
N2-isomers (cycle B) |
 |
|
|
|
Using method of
potentiometric titration by 0.1 N
NaOH in 50% methanol – 50% 0.1 N aqueous solution NaNO3
constants of acidity were determined.
|
|
n |
pKa
(25 ˚C) |
|
OH-acids
|
NH-acids |
||
|
N1-isomers
(cycle B) |
1 |
2.96±0.04 |
3.03±0.06 |
|
2 |
3.01±0.05 |
3.01±0.05 |
|
|
3 |
2.95±0.05 |
3.06±0.06 |
|
|
N2-isomers
(cycle B) |
1 |
2.91±0.06 |
2.93±0.05 |
|
2 |
3.01±0.06 |
3.01±0.05 |
|
|
3 |
3.09±0.05 |
3.15±0.06 |
|
As expected, values pKa of the NH-unsubstituted tetrazoles differ from pKa of the corresponding carboxylic acids a little. The differences in рКа between N1- and N2-isomers
are not essential too.
Shadows and
Intersections of RNA secondary structures
Ulrike Mückstein and Kurt Grünberger
Institute for Theoretical Chemistry and Structural Biology, University
of Vienna, Währingerstraße 17, A-1090 Vienna, Austria
Many RNA sequences fold into the same secondary structure. Two sequences that share the same shape are termed "neutral" with respect to their structure. Neutral
sequences that have a common secondary structure form an extensive mutationally connected network. We call such networks "neutral networks". The neutral network of a
common structure is densely embedded in the set of sequences that are compatible with this structure. Therefore the neutral networks of two common structures come very
close together on their intersection (i.e. the set of
sequences that are compatible with both structures).
The evolutionary dynamics of RNA populations can be investigated by computer simulations. A trajectory of a typical simulation experiment in a flow reactor describes the time
course of the distance of the population average from a predefined target secondary structure. Neutral networks convey an ideal combination of search capacity and robustness
to mutations in such experiments: the genotypes (sequences) may diffuse over the network by single nucleotide exchanges without loosing the currently optimal structure, until a
non neutral mutant, whose structure is closer to the target, is encountered. The population will then switch to the network of this structure. In order to be of maximal use, neutral
networks should be connected
graphs and a maximal number of new structures should be available in the
1-error neighborhood of the network or even a single sequence.
The set of all possible secondary structures that appear in the 1-error neighborhood of the neutral network of a given structure is termed its "shadow". A "local shadow" can be
generated from a stochastic selection of sequences of a neutral network by determining the structures in their 1-error neighborhoods. The intersection of different local shadows
of the same structure comprises the set of the most common
neighbor of this structure.
In this contribution we use shadows and intersections of secondary structures to study accessibility relations in sequence space. Deeper insights into the accessibility relation
between RNA structures permit a better understanding of local events like cycles of recurrent structures and the emergence of different kinds of transitions between structures
in
evolutionary trajectories.
Analysis and planing of experiments with enzyme
inhibitors
Irena Mudri 1, Slobodan Rendić 2, and Paško Konjevoda 3
1PLIVA Research Institute, Prilaz Baruna Filipovića 29, Zagreb, Croatia
2Faculty of Pharmacy and Biochemistry, Ante Kovačića 1, Zagreb, Croatia
3Ruđer Bošković Institute,
Bijenička 54, Zagreb, Croatia
Enzyme inhibitors are substances that inhibit the catalytic activity of an enzyme. Enzyme inhibition can the desirable terapeutic effect. Also, it can be the side-effect of
interaction with different enzyme systems, including cytohrome P-450 family. In this paper we analyse the statistical quality of the papers describing the interaction of
different substances with cytohrome P-450 family. The majority of the analysed papers shows the serious weaknesses in analysis and presentation of experiments.
The most common are:
(a) the use of the linear
models of enzyme kinetics in situations where non-linear models are more
appropriate;
(b) insufficient number of
measurements;
(c) total lack of information
about the numbers of measurements;
(d) violated assumptions about heteroskedasticity;
(e) inadequate description of the measures of the variation (the standard error vs. the standard description);
(f) total
lack of the information about statistical tests for the difference in the
slopes and interceptors;
(g) inadequate power analysis
in the planning of the experiments.
Our conclusion is that the largest part of published papers can not be used as a reliable source of the information for the comparison of different substances. We
also propose the minimal standards for publishing of enzyme kinetics data.
A Minimal Model for Gene Regulatory Networks
Stefan Müller, Christoph Flamm,
and Peter Schuster
Institute for Theoretical Chemistry and Structural Biology, University
of Vienna, Währingerstraße 17, 1090 Wien, Austria
We developed a generic model of a minimal cell for investigating evolutionary and developmental questions. One major part of the model is a gene regulatory
network (GRN) which allows to study the dynamics of gene expression.
Our
model uses the following abstraction of the chemical processes within a living
cell:
(1)
The system is closed (mass conservation).
(2)
Gene products bind to the regulatory regions of the genes and either enhance or
inhibit their expression.
(3) Polymers
(proteins, RNAs) are built from activated monomers.
(4)
Transcription and translation are one-step reactions.
(5)
Polymers degrade yielding deactivated monomers.
(6)
The system is coupled to the environment via a photochemical reaction which
transforms deactivated into activated monomers.
(7)
Proteins can catalyze the activation of the monomers.
The topology of the GRN is defined in terms of an edge-weighted directed graph with vertices representing genes, edges representing gene-protein binding and edge
weights representing binding constants and either enhancement or inhibition. This graph can be "decoded" from an artificial genome. Evolutionary operators like mutation and
recombination act at the level of the artificial genome and cause changes in the GRN via an intricate decoding process, but always yield a valid GRN. Therefore evolutionary
questions can be easily addressed within this framework.Alternatively, the GRN can be designed explicitly. Such rational network design may lead both to the engineering of
new cellular behaviors (e.g. the repressilator) and to an
improved understanding of naturally occurring networks.
chemical equations in the minimal cell which is finally mapped to a system of ordinary differential equations (ODEs). Integration of the ODEs yields the gene expression
pattern over time.
Summary:
(1) The model is generic.
(2) The model is
"exact" chemically and computationally.
(3) The model is a
"construction kit" for GRNs.
(4) The model displays a
variety of dynamical behavior.
QSAR Study of Phthalimide Derivatives
Sonja Nikolić1, Marica Medić-Šarić2, Julija Matijević-Sosa2, and M. Puzović3
1Ruđer Bošković Institute, HR-10002
Zagreb, POB 180, Croatia
2Faculty of Pharmacy and Biochemistry,
University of Zagreb, Zagreb, Croatia
3Croatian Institute of Transfusion Medicine, Zagreb, Croatia
Various phthalimide derivatives have been known for a long time as substances with different biological activities as cytostatic, plant growth regulators, bacteriostatic and
fungistatic. The interest for investigation of these substances has increased after the discovery of teratogenic activities of thalidomide (a-phthalimidoglutarimide), used as
sedative. Thalidomide showed inhibition of angiogenesis and is being evaluated as an anticancer agent and drug for diabetic retinopathy and osteoporosis.
On the basis of suppression of inflamation and immunomodulation thalidomide is in use as drug for leprosy (ENL) already from 1964 and now is investigated for AIDS.
The dermatological effects are great and some of the results in vitro appears that thalidomide may be able to inhibit HIV replication.1
Structure-activity relationships of thalidomide and analogs showed that some of the simple derivatives, as N-OH-phthalimide, were more effective and it is therefore the
reason for further investigation of phthalimide derivatives.
In our work we have studied mitodepressive activity, %I, of some phphalimides with isoxazolone or pyrazolone rings, hydroxamic acids and other substituents. Their
partition coefficients, log P (o/w), were calculated according to Rekker method By using the methods of molecular modeling, the relationships between structure, properties
and activity of these compounds were investigated. Three topological indices were used: Wiener index, the valence connectivity index, and the Balaban index. 2
1. A.L. Moreira, L.G. Corral, W. Ye, B. Johnson, D.
Stirling, G.W. Muller, V.H. Freedmann, G. Kaplan (1997) AIDS Res. Hum. Retroviruses 13:
657-663.
2. M. Vedrina, S. Marković, M. Medić;-Šarić, N. Trinajstić (1997) Computer Chem. 21: 355.361.
Algorithms
for drawing polyhedra from 3‑connected planar graphs
Alen Orbanić, Marko Boben, Gašper Jaklič, and Tomaž Pisanski
IMFM, University of Ljubljana, Jadranska 19, SI-1000 Ljubljana,
Slovenia
Two algorithms for producing
polyhedral representations for 3-connected planar graphs are
discussed in the paper. One of them uses Tutte's drawing algorithm to produce
2D drawing.
Then the drawing is lifted into 3D space obtaining a polyhedral embedding.
The other is simple algorithm by G. Hart1 for drawing canonical
polyhedral representations. Some alternative aspects (physical model, Markov
chain model) in algorithms
for obtaining Tutte's drawings are presented and proved. The underlaying graphs of some chemical structures (Fullerenes, ...) are 3-connected planar graphs.
1. Hart G.W. (1997) Calculating Canonical Polyhedra, Mathematica
in Education and Research 6 (3):
pp. 5‑10.
NEW HORIZON OF TETRAZOL CHEMISTRY
Vladimir A. Ostrovskii
Saint Petersburg State Institute of Technology (Technical University), 26,
Moskovsky prosp., Saint Petersburg, 190013, Russia
The prognosis of successful application of tetrazoles in modern medicine and engineering given in the review1 proved to be correct. The medicines of a new generation
containing tetrazole ring
in a molecule such as Lozartan are released to
world pharmaceutical market.

Tetrazoles
are used as the components of gas‑generating formulations, agents of
metals protection from corrosion, modern photographic materials, etc.
For the last five years the significant successes in development of effective methods of tetrazole synthesis are achieved. Today the directed tetrazoles production sold both in
laboratory and in industrial scales is became possible.
Traditionaly, the scientific schools of Russia and Belorussia significantly contribute to the development of tetrazole chemistry. In the begining of new century the interest to the
given problem of the world leaders, such as K. Sharpless (Nobel award in
2001) is also observed.2
As a result of Russian and foreign scientific schools efforts association the advance of tetrazoles on pharmaceutical and other perspective markets of chemical products is
provided in a near future.
1. G.I. Koldobskii,
V.A. Ostrovskii (1994) Usp. Khimii 63: 847.
2. Z.P. Demko,
K.B. Sharpless (2001) J. Org. Chem. 66:
945.
The Nest/Egg Motif in Proteins
Debnath Pal1, Manfred S. Weiss2, and Jürgen Sühnel3
1Molecular Biology Institute, Universty of
California, Los Angeles, CA, USA
2EMBL Hamburg Outstation, Hamburg,Germany
3Institute of Molecular Biotechnology, Jena
Centre for Bioinformatics, Jena, Germany
The main-chain conformations of protein chains can be conveniently analyzed by means of the Ramachandran map displaying the distribution of amino acid backbone torsion
angles π and ψ .1 The interpretation of Ramachandran maps usually focuses on clusters for specific secondary structure elements such as β‑sheets and different helix types. In
these cases the main‑chain conformations of successive
amino acid pairs are identical or at least similar.
Recently, Watson and Milner-White2 have discovered that many anion and cation binding sites in proteins are made up by a sequence of three amino acids of which two exhibit
"enantiomeric" main‑chain conformations. Here, contrary to the conformations in β‑sheets and helices, the main‑chain torsion angles (π,ψ) of the two adjacent amino acids are
approximately inverted about the center of the Ramachandran map. The authors have called this motif a nest because three successive residues obeying this torsion angle
criterion form a concave depression. In the majority of cases, the nests do bind to an atom or a group of atoms, which we suggest may, as a binding partner of a "nest", be
descriptively and conveniently called "egg".3 It is intriguing that many structural motifs described previously, such as Schellman loops, the oxyanion holes of serine proteases,
P‑loops in ATP- or GTP‑binding proteins can be subsumed under this nest/egg-concept. Watson and Milner-White conducted their analysis on a limited database of 67 protein
structures. We have performed an extended analysis of their concept in a database about 20 times larger3 (the same database as the one used in ref. 4). It turns out that some of
the results obtained by Watson and Milner‑White are confirmed, others have to be modified and some new aspects are unveiled as well. For example, detailed information on the
amino acid occurrence in nests and the nest occurrence in secondary structure elements is reported and a thorough analysis of the egg occupancy of nests is presented. The
nest/egg concept described here takes ligands, co‑factors, water molecules, etc. into account and therefore sets the stage to a general approach to binding sites in proteins. It
should also be of importance to other aspects of protein structure analysis and prediction. A preliminary analysis has shown that the nest motif is only one example of a more
comprehensive list of non‑repetitive main‑chain dipeptide conformations. Finally, one may speculate whether these motifs may constitute stable structures very early along the
pathway of folding since they are inherently local structures and thus would not require a large entropy reduction upon formation. 1. Ramachandran G.N., Sasisekharan V. (1968) Adv. Prot. Chem. 23: 293. 2. Watson J.D., Milner-White E.J. (2002) J. Mol. Biol. 315: 187-198,199-207. 3. Pal D., Sühnel J., Weiss M.S. (2002) Angew. Chem. Int. Ed. 41: 4663.4. Brandl M. et al. (2001) J. Mol. Biol. 307: 357-377.
Drawing the resonance graphs of catacondensed
benzenoid graphs
Igor Pesek and Aleksandar
Vesel
Department of Mathematics, PEF, University of Maribor, Koroška c. 160,
SI-2000 Maribor, Slovenia
The vertices of the resonance graph R(G) of a benzenoid graph G are the Kekulé structures (or 1-factors) of G. Two vertices of R(G) are adjacent if the corresponding
Kekulé structures interact, that is if they differ in the position of just three double bonds. An algorithm for drawing the resonance graphs of catacondensed benzenoid graphs
using the
canonical binary coding of 1‑factors has been developed. Additionally,
the algorithm was implemented in a software package for visualizing this class
of graphs.
Anti-connectivity in QSAR studies
Matevž Pompe1 and Milan Randić2
1University of Ljubljana, Faculty of
Chemistry and Chemical Technology, Aškerčeva 5, SI‑1000 Ljubljana,
Slovenia
2National Institute of Chemistry, Hajdrihova
19, SI-1000 Ljubljana, Slovenia
Several properties exists where presence of certain atom or functional group suppresses the signal. For instance, the presence of heteroatom in the molecule has negative
effect on the response factor of FID detector in gas chromatography. Similar observation could be found in modeling biological activities and reaction rate constants. None of
the classical bond additive or atom additive topological indices has the ability to describe such behavior, this also include present state of the art variable topological indices.
A new procedure for the calculation of
variable connectivity indices will be presented, which enables modeling of the
described anti-connectivity properties.
Electron reorganization in chemical reactions.
Structural changes from the analysis of bond order profiles
Robert Ponec, Gleb Yuzhakov, and David L. Cooper
Institute of Chemical Process Fundamentals, Czech Academy of Sciences,
Rozvovova 135, Prague 6, CZ 165 02, Czech Republic
Bond orders represent the theoretical counterpart of the classical concept of bond multiplicity, and their variation along the reaction path allows one to visualize the abstract
"electron reorganization" in terms close to classical chemical thinking. Due to this ability to get a detailed insight into the electron reorganization during the chemical reactions
these dependencies, known as bond order (BO) profiles were proposed and applied as a new tool for the study of chemical reactivity. Despite broad use of these profiles,
several important questions concerning their applicability and mechanistic interpretation still remained to be answered. These questions concern especially the following
problems:
i. the sensitivity to the particular
choice of the definition of bond order
ii. the sensitivity to the quality of the computational method used
iii. the possibility of the
mechanistic interpretation of bond order profiles in terms of Hammond postulate.
The Effect of a Certain Activity on
Triglyceride and Cholesterol Blood Serum
Mohammad Javad Pourvaghar
Department of Physical Education, Faculty of Science, University of
Kashan, Ravand Boulevard, 87317‑51167 Kashan, Isfahan, Iran
In this research we use scientific and experimental methods. In this study, 15 students participated. They were students of physical education. The purpose of this study was
to find out changes of cholesterol and triglyceride in their blood serum after aerobic and anaerobic activities. Blood samples were used to measure cholesterol and
triglyceride, one sample before the activity and other after it. The results showed a decrease in the cholesterol level and increase in the triglyceride level of their blood.
The observed difference between the two samples for triglyceride was significant (observed t = -3.28). However, this difference for cholesterol was non-significant
(observed t = ‑2.28). This shows that aerobic and anaerobic activities have influence on the level of triglyceride. This can be due to the time of physical exercise secretion
of catecholamines which facilitates release of free fatty acid (FFA) from body tissues. Also, the result indicated that aerobic and anaerobic activities have no influence on the
cholesterol in the blood. This can be explained in terms of
the normal of cholesterol of the participants.
Relative Rate Tests for Conserved Non-Coding
Sequences
Sonja Prohaska
Bioinformatics Group, Institute of Comupter Science, University of
Leipzig, Kreuzstraße 7b, D‑04103 Leipzig, Germany
In many eukaryotic genomes only a small fraction of DNA codes for proteins. Nevertheless, geneticists focused on the evolution of coding sequences for a long time. The
knowledge about evolutionary mechanisms is therefore deduced from coding sequences and the methods for testing the molecular evolutionary clock hypothesis were
developed for the application to genes and protein sequences. But there are a lot of important genetic elements in the larger fraction of non-protein coding DNA which direct
the development and the physiology of the organisms. Such elements are, for example, promoters, enhancer, insulators and micro-RNA genes. The molecular evolution of
these elements
is difficult to study because their functional significance is hard to deduce
from sequence information alone.
Here we present an approach to detect rate heterogeneity in the evolution of putative cis‑regulatory elements at a macro-evolutionary scale: Conserved Non-Coding
Nucleotide (CNCN) sequences are identified from the comparison of two outgroup species by the use of phylogenetic footprinting. These CNCN sequences are then
compared to their homologous sequences in a pair of ingroup species. The degree of modification these sequences suffered in the two ingroup lineages are then tested for
rate differences independently from the estimation
of modification rates.
We apply this method to the full sequences of the HoxA clusters from six gnathostome species: a shark (Heterodontus francisci), a basal ray finned fish
(Polypterus senegalus), an amphibian (Xenopus tropicalis) as well as three mammalian species, human, rat and mouse. The results show that the evolution rate of
CNCN sequences is not distinguishable among the three mammalian lineages, while the Xenopus lineage has a significantly increased rate of evolution. Furthermore
the estimates of the rate parameters suggest that in the stem lineage of mammals the rate of CNCN sequence evolution was more than twice the rate observed within the
placental mammal clade, suggesting a high rate of evolution of cis-regulatory elements during the origin of amniotes and mammals. We conclude that the proposed methods
can be used for testing hypotheses about the
rate and pattern of evolution of putative cis-regulatory elements.
joint work with: Guenter P. Wagner,
Claudia Fried, and Peter F. Stadler
Novel Graphical Representations of DNA Sequences as a Map
Milan Randić
National Institute of Chemistry, Hajdrihova
19, POB 3430, SI-1001 Ljubljana, Slovenia
In early and mid 1990s notion of graphical representation of lengthy DNA sequences was introduces by Hamori, Morgenthaler, Nandy and others. Novel graphical
representations include modified 2-D representations and a 3-D representation of DNA – all that can be associated with matrices used for numerical characterization of
graphical forms of DNA. Most recent development have lead to representation of DNA as a 2-D map either by adopting a binary codes for the four nucleic acid bases or
using a spiral format over Cartesain coordinate system to form numerous local regions associated with the four nucleic acids bases. We will describe properties of these
novel graphical representations of DNA. An important advantage of map‑representation of DNA sequences is their “compact” format, which allows long DNA sequences to
be presented graphically on a relatively small space but still preserving most of the details of the representations.
Universal Languages of the Past and the
Present
Milan Randić
3225 Kingman Rd, Ames, IA 50014, USA
We will start with a brief review of attempts in the 17th and 18th centuries to create universal languages, in particular giving some details on the work of Mersenne, Giordano
Bruno and Leibniz. We will also briefly outline work of Bliss in 1960-1980s. NOBEL – Universal pictographic language of this author, as a potential universal language of the
present - will be introduced by showing basic 120 signs to be followed with half a dozen operational rules for construction of numerous additional words related to the basic
sings. These rules include “doubling” and “tripling” of the signs, “overlapping of signs,” “crossing” and “inversion” of sings, construction of “reciprocals” and “fraction” sings.
With all these construction alternatives it was possible to construct NOBEL Dictionary with some 15,000 words, a sample section of which will be illustrated. We will end
presentation with illustration of selection of texts written in NOBEL.
Novel
Graphical Matrix and Distance‑Based Molecular Descriptors
Milan Randić1, Nabamita Basak2, and Dejan Plavšić3
1Natural
Resources Research Institute, University of Minnesota Duluth, 5013 Miller Trunk
Highway, Duluth, Minnesota 55811, USA
2Department
of Chemistry, University of Minnesota Duluth, 10 University Drive, Duluth,
Minnesota 55812, USA
3Ruđer
Bošković Institute,POB 180, HR-10002 Zagreb, Croatia
We draw attention to graphical matrices as a source of numerous structural invariants that could be used as predictor variables in QSPR and QSAR studies of molecules. In
particular we put forward a novel graphical matrix G associated with a molecule whose element [G]ij is the subgraph of the corresponding molecular graph obtained from it
by deleting vertices i and j. Several molecular descriptors have been extracted from the G matrix and its numerical realization, the Gw matrix, based on the Wiener index.
The usability of the extracted "double" invariants as predictor variables in QSPR studies of molecules has been tested on the total steric energies of octane isomers.
Estimation of Stability of Coordination Compounds by Overlapping
Spheres Method
Nenad Raos
Institute for Medical Research and Occupational Health, HR-10001
Zagreb, P.O.B. 291, Croatia
The method of overlapping spheres was applied for the estimation of stability constants of copper(II) chelates with N-alkylated glycines. The central sphere with radius 0.3 or
0.4 nm was situated at the center, vertices or faces of the coordinaton octahedron. The stability constants for mono- (log β110) and bis-complexes (log β120) of glycine and
its methyl, dimethyl, ethyl, diethyl, propyl, butyl, iso-propyl and tert-butyl derivatives were correlated on the overlapping volume of the central sphere and the van der Waals
volumes of the neighbouring atoms. The calculations were performed with the two independent sets of stability constants (N = 6, 8 and 5, 8 for mono- and bis-complexes,
respectively).
The better correlations were obtained for mono- (r = 0.768 – 0.968) than for bis-complexes (r = 0.737 – 0.922). The exclusion of complexes with N‑iso‑propylglycine and
N‑tert‑butylglycine yielded substantially better results (r = 0.741 – 0.997 for mono‑complexes, r = 0.877 – 0.984 for bis-complexes). The best positions of the central
sphere proved to be the faces of the coordination octahedron (r = 0.909 – 0.985 for mono‑complexes, r = 0.843 – 0.984 for bis-complexes), and the worst potitions
appeared to be that of the ligated oxygen atoms (r = 0.741 – 0.979 and 0.737 – 0.953 for mono- and bis‑complexes, respectively). The best regression lines reproduce
stability constants with the error 0.00 - 0.2 log β units, except the constants of N-iso-propylglycine and N‑tert‑butylglycine chelates, which were estimated with a typical
error around 0.5 log β
units.
Managing the Complexity of Biomedical
Information
Jörg Reichardt1, Stefan Bornholdt1, and Peter Ahnert2,3
1Interdisciplinary Center for Bioinformatics,
University of Leipzig, Kreuzstraße 7b, D‑04103, Leipzig, Germany
2Center for Biotechnology and Biomedicine,
Junior Research Group „Molecular Diagnostics – Microarray Techniques“,
University of Leipzig, Leipzig, Germany
3Institute of Clinical Immunology and
Transfusion Medicine, Faculty of Medicine, University of Leipzig, Leipzig,
Germany
In the life sciences vast amounts of information are being accumulated worldwide. They are stored partly as machine readable databases and largely as plain text, as
catalogued in the PubMed database. These data represent a formidable resource of biomedical knowledge. Formidable, however, is also the task to exploit this wealth of
information, e.g. to obtain an overview of all aspects pertaining to a particular question. Few tools are available so far to condense large amounts of information into
meaningful and preferably abstracted entities.
In order to facilitate the management of large amounts of data, we introduce a new clustering algorithm that can operate on any kind of relational data represented as a
network. The algorithm is based on a q-state Potts model and allows for fuzzy clusters. Clusters are found as domains of equal spin value in the ground state of a modified
Hamiltonian. No prior knowledge of the number of clusters has to be assumed. Measuring affiliation ratios of nodes to clusters can account for overlapping clusters and
quantify the association strength of nodes to clusters as well as the stability of a cluster.
Together with three other programs, this algorithm was implemented in the KEN series of tools. It was tested in an analysis of a gene-gene co-citation database of PubMed
obtained from text-mining. The algorithm mines the structure of the co-citation network to identify de novo clusters of biologically relevant information, e.g. groups of highly
connected genes. Statistical analysis of the annotation of the genes in these clusters allows for the identification of research themes in the topic under study. Results of the
analysis, its
strengths and limits, are presented and discussed.
J. Reichardt, S. Bornholdt,
arXiv:cond-mat/0402349.
T. J. Jenssen, A. Laengreid,
J. Komorowski, E. Hovig (2001) Nature
Genetics 28: 21.
B. Muetzel, H.H. Do, K.
Pruefer, E. Rahm, S. Paabo, in
preparation.
H.H. Do,
E. Rahm (2004) Proc. EDBT 2004,
Heraklion, Greece, Springer LNCS.
From Dendrograms to Topology
Guillermo Restrepo and José L. Villaveces
Departamento de Química, Facultad de Ciencias Básicas, Universidad de Pamplona, Pamplona, Km 1 via Bucaramanga, Colombia
We developed a mathematical methodology to study sets of objects of chemical interest using cluster analysis and topology. We defined each chemical object by a point in
a space of properties built up using physico-chemical, graph-theoretical and quantum chemical properties of the elements of each set. After, we apply cluster analysis on the
set of chemical interest Q and finally we obtain dendrograms. Based on such trees (dendrograms), we carried on a topological study of Q introducing the concept of
neighbourhood. Thus, we defined a basis for a topology for Q using the neighbourhoods mentioned above. Finally, we studied some topological properties such as the
closure and the boundaries of particular subsets of Q. Besides, we show four practical examples of the methodology studying 72 chemical elements, 31 steroids, 238
benzimidazoles and 20 amino acids. Among results obtained is important to remark that the mathematical boundary of metals and non-metals are the same elements, which
are semimetals. On the other hand in set of steroids it is possible to establish a partition in disjunct subsets which result intuitive ones according to structural and chemical
reactivities ideas. Finally, we found in sets of benzimidazoles and amino acids that there are some substances that are between some particular sets (boundary substances).
Static and dynamic properties of branched
polymers
Piotr Romiszowski and Andrzej Sikorski
Department of Chemistry, University of Warsaw, Warsaw, Poland
Monte Carlo simulations of lattice star-branched polymers in diluted solution confined between two parallel impenetrable walls were performed. The sampling of the
conformational space was done by means of the local micromodifications of the chain conformation. The static and dynamic properties of the system have shown that the
distance between the walls has an influence on the dimensions of the molecules. The effect of a deformation of the molecule by the walls has been observed and discussed,
especially the possible changes in the mechanism of chain motion have been pointed out. Also, the influence of the distance between the walls on the short‑time dynamics as
well as long‑time dynamics have been investigated and discussed. The impact of the introduction of impenetrable obstacles into the system on chain’s properties was also
studied.
Computer simulation of polypeptide motion
Piotr Romiszowski and Andrzej Sikorski
Department of Chemistry, University of Warsaw, Warsaw, Poland
In the present work we studied a simple model of a polypeptide chain in the confinement. The model chain was restricted to a flexible [310] lattice and were represented as a
sequence of united atoms located at the positions of α carbons. The force field introduced into the model consisted of the long-range contact potential between amino acid
residues with the excluded volume and a local preference of forming helical states. The chain was built of two kinds of residues: hydrophilic and hydrophobic. The properties
of such chains were determined by means of the Monte Carlo simulations. During the simulations we observed and monitored the translocation of the chain during its passing
through a hole in an impenetrable wall. The influence of the chain length, the sequence of residues and the temperature of the system on the structure of the chain during the
translocation was investigated. The dynamic properties of the system such as the mean first passage time were studied and discussed.
Quantum Chemical Parameters
for Structure‑Activity Relationships – Mechanistic Interpretation and
Dependence on the Level of Theory
Gerrit Schüürmann
Department of Chemical Ecotoxicology, UFZ Centre for Environmental
Research, Permosersrtaße 15, 04318 Leipzig, Germany
Quantum chemistry offers a mechanistic route to characterize molecular structures with respect to geometric and electronic features that govern their biochemical reactivity
and associated biological activity. So far, however, little attention has been paid to the dependence of descriptor values on the level of theory. This holds true in particular
with respect to potential discrepancies between semiempirical and ab initio methods when quantifying measures for the electrophilicity and nucleophilicity as well as for
the molecular charge distribution. In the present paper, the background, mechanistic meaning and method dependence of common quantum chemical reactivity parameters
are discussed. Taking a data set of more than 600 organic compounds, descriptors based on frontier orbital energies, molecular wavefunctions, surface areas as well as on
the electronic charge distribution are comparatively analyzed using three semiempirical and two ab initio quantum chemical methods. For certain types of parameters and
chemical functionalities, the results reveal systematic differences, particularly as regards the characterization of the electronic charge distribution in terms of net atomic
charges. The discussion includes examples of QSAR applications employing various quantum chemical descriptors.
DFT Calculation of NMR CSA Tensors
Vilko Smrečki1 and
Norbert Müller2
1 Ruđer Bošković
Institute, NMR Center,
Bijenička 54, POB 180, HR-10002 Zagreb, Croatia
2 Institute of Chemistry,
Johannes Kepler University, Altenbergerstr. 69, A-4040, Linz, Austria
Density Functional Theory (DFT) calculations were used to quantitatively predict the influence of protein secondary structure elements (F and Y angles) on NMR chemical
shielding anisotropy (CSA)
tensors as a prerequisite to structural interpretation of experimental CSA
values.
The Gaussian98 program package was used employing the 6‑311G(d,p) basis set for geometry optimization and 6‑311++G(3df,3pd) basis set for NMR parameters
calculation. The Becke's three parameter hybrid functional with the Lee, Yang and Parr correlation functional (B3LYP) was employed in DFT calculation. The
gauge‑independence requirement for NMR parameter
calculation was treated with the Gauge‑Invariant Atomic Orbital (GIAO)
approach.
The molecular models used in the
simulation of protein secondary structure elements and CSA calculations were
capped alanine dipeptide, tripeptide and octapeptide.
The results of DFT calculations of 1H, 13C and 15N CSA tensors showing dependence on secondary protein structure elements will be demonstrated by way of
Ramachandran-CSA
diagrams for the dipeptide model.
Acknowledgement
This work was supported in part by the Austrian Science Fund (FWF) Lise Meitner fellowship (M677), FWF project P15380, Austrian‑Croatian joint research project
911-02/03-06, and the Ministry of Science and Technology
Republic of Croatia project P0098059.
Gene Phylogenies of non-coding RNAs
Peter F. Stadler
Bioinformatics Group, Department of Computer Science & Interdisciplinary Center of Bioinformatics,
University of Leipzig, Kreuzstraße
7b, D-04103, Leipzig, Germany
Non-protein-coding RNAs (ncRNAs) are known to play significant roles in biological systems. They belong to numerous very diverse classes, even within one class they
share few or no common sequence motifs, they are often to short to be characterized based on secondary structure alone, and they occur in a wide variety of oftentimes
poorly understood genomic context.Some of these groups, for instance rRNAs, or tRNAs are known to be ancient, their origins predating the last common ancestor of all
living organisms. Other classes, such as vertebrate telomerase RNA, is a recent addition to cell RNA repertoire. A major obstacle in investigating non-coding RNAs is
their relatively small size compared to protein-coding genes. Furthermore, we are still lacking reliable computational tools for their detection, so that only families with
relatively well-conserved sequences can be readily annotated in newly sequenced genomes.
The evolutionary origins and history of the non-coding RNAs has not received much attention, maybe as a consequence of these technical difficulties. Recent work,
however, indicates that some classes of non‑coding RNAs exhibit a complex history governed by individual gene duplication and a later series of duplications that appears to
be associated with major radiations.
We will discuss here methodological issues concerning both the detection of non-coding RNAs and the elucidation of their evolutionary relationships. As examples we
describe the evolutionary history of the mir17 and mir23 microRNA clusters, we consider the co-evolution of several microRNAs with Hox gene clusters and we discuss
first results concerning the evolution of certain groups of snoRNAs.
joint work with: Andrea Tanzer (Leipzig & Vienna) and Bettina Müller (Leipzig and München/Weihenstephan)
Molecular Evolution of a MicroRNA Cluster (2004) J.Mol.Biol. 339: 327-335.Evolving towards the hypercycle
Camille Stephan-Otto Attolini and Peter F. Stadler
Theoretical Biochemistry Group, Institute for Theoretical Chemistry,
University of Vienna, Währinger Straße 17, A-1090 Vienna, Austria
Following some ideas about prebiotic chemistry and origin of life theories, we have combined the use of RNA-secondary structures as a representation of the phenotype of
RNA molecules, and the capability of some of these molecules to self-replicate and catalyze other's replication.
A model of interacting RNA molecules is presented, where replication and catalisys is assumed for every molecule. A number of secondary structures are used as targets
which are organized in a hypercycle. This set is approached by the sequences via mutation and selection. Replication and catalytic rates depend inversely on the distance to
the target. The system is embeded in a two-dimensional lattice where replication and diffusion take place. The topology of the targets' set is also modificated in order to
model the behaviour of the sequences when short-cut or selfish parasites are present.
In former studies, this kind of systems resulted unstable against the two of them. In our model, spatial patterns and evolution toward the targets, give the system resistance
against all kind of parasites. A quasispecies behaviour is observed in the sequence distribution and diffusion in sequence space is shown to occur and to be related to the
mutation rate.
Alignments of Partly Coding Regions: The code2aln - Project
Roman R. Stocsits1, Ivo L. Hofacker2, and Peter F. Stadler1
1Bioinformatics Group, Department of Computer Sciences & Interdisciplinary Center of Bioinformatics,
University of Leipzig, Kreuzstraße 7b, D-04103 Leipzig, Germany
2Institute
of Theoretical Chemistry & Structural Biology, University of Vienna,
Währingerstraße 17, A-1090, Vienna, Austria
High quality sequence alignments of RNA and DNA sequences are a prerequisite for the comparative analysis of genomic sequence data. The high level of sequence
heterogeneity, as compared to proteins, makes good alignments of nucleic acid sequences often impossible. In many cases, the nucleic acid sequences under consideration, or
parts of them, code for proteins. While protein sequences can still show substantial homology, the corresponding nucleic acid sequences have already evolutionarily diverged,
thus they are essentially randomized. This is caused by the inherent redundancy of the genetic code: Most amino acids have more than one codon on the level of nucleic acid.
This specific problem leads to gaps and incorrectly aligned segments within coding regions.We implemented a multiple nucleic acid alignment procedure that uses genetic information about coding and non-coding regions as part of the scoring function in order to
improve the resulting alignment. Our algorithm combines (mis)match scores for nucleic acids with those for the underlying amino acids in the case of open reading frames and
exons. The program makes explicit use of information about overlapping open reading frames, as they occur in virus sequences, to further improve the reliability and quality of
the nucleic acid alignment.
An application to two unrelated groups of viruses is described. We processed the alignments as input for a procedure for detecting conserved RNA secondary structure
elements in RNA genomes of Leviviridae and the pregenomic RNA of human hepatitis B virus. Virus genomes contain various (partially overlapping) open reading frames and
are an ideal test case for a procedure that makes usage of information about (overlapping) coding regions to improve the input alignments and, therefore, the identification of
conserved secondary structure elements. We find a number of highly significant secondary structure elements, not being described in the literature so far, and some well known
elements like the epsilon-elements and two important elements of the HPRE region in hepatitis B virus. Also the results of the Levivirus group are of particular interest: We
detect various secondary structure elements that are strongly confirmed by compensatory mutations and gain novel insight into the structural organization of Levivirus genomes.
New Insights into Protein Structure from
Structural Bioinformatics Approaches
Jürgen Sühnel
Biocomputing Group, Institute of Molecular Biotechnology, Jena Centre
for Bioinformatics, D-07745 Jena,
Germany
Despite much work and a database of experimentally determined biopolymer structures that is increasing frighteningly fast, success in the prediction of three-dimensional
(3D) structures of biological macromolecules is scarce. One explanation may be that the currently known structural principles do not disclose the complete picture and that
new concepts and new ideas are necessary to propel the field from the mainly descriptive into a more predictive mode. Structural bioinformatics approaches try to obtain
new biological knowledge by statistical analyses of 3D biopolymer structure data. In the talk I will report on the possible role of CH...p-interactions in proteins1 and on a
generalized concept of hydrogen bonding that is emerging from new results obtained by others as well as by our group.2 More recently, we have worked on the so-called
nest/egg motif in proteins3 that was first described by Watson and Milner-White in the beginning of 2002 and that is, contrary to the situation within α-helices or ß-sheets,
characterized by dramatic backbone torsion angle changes between sequence neighbours. This corresponds to transitions between the allowed clusters in the Ramachandran
map. We now call these amino acid pairs non‑repetitive dipeptides. Very recently, we have discovered (unpublished work) that the nest motif is only a special case of this
dipeptide type and that on average 25% of all dipeptides in proteins are of the non-repetitive type. For individual proteins this fraction may vary between 0 and even 60%.
We believe that these new findings have led us to a new vista point from which we can have a fresh look at protein structure with exciting new prospects for structure
validation and prediction and hopefully also for protein evolution.
1. Brandl M. et al. (2001) J. Mol. Biol. 307: 357.
2. Weiss M.S. et al. (2001) Trends Biochem. Sci. 26: 521.
3. Pal D. et al. (2002) Angew. Chem. Int. Ed. 41: 4663.
Exactly Solvable Models for Investigation of
Dynamical
Alina A. Suzko
Laboratory of Information Technologies, Joint Institute for Nuclear Research, 41980 Dubna, Russia
A method of constructing a time-dependent amiltonian is elaborated for which a system of Schrődinger equations and Pauli equations admits analytic solutions. Time‑independent
soluble problems are transformed into time‑dependent ones by a set of unitary time‑dependent transformations and a proper choice of initial states. In particular, the periodic
time‑dependent Hamiltonians are generated whose expectation values for cyclic solutions and spin‑expectation values do not depend on time. This approach can be used for
modelling quantum dynamic wells with the effect of the particle localization.
When studying the composite systems possessing both external and internal degrees of freedom, it is necessary to take into account the mutual influence of slowly varying
external fields and rapidly varying intrinsic fields. The generalized technique of Darboux transformations and the inverse scattering problem is worked out for reconstructing
time‑dependent and time‑independent potentials and their corresponding solutions in a closed analytic form on the basis of the inverse scattering problem in the adiabatic
representation. Matrix elements of an exchange interaction determining the slow subsystem Hamiltonian are calculated and studied in terms of exact solutions obtained within
the arametric inverse problem on the semi-axis and on the entire axis. The influence of parametric spectral characteristics of the fast subsystem on the behaviour of the slow
subsystem is investigated. It is shown that the particularities of the slow subsystem Hamiltonian are determined by the nature of the parametric fast Hamiltonian: namely, the fast
subsystem is given on the semi-axis or on the entire axis. In particular, it is shown that in the case of the parametric problem on the entire axis potentials, eigenfunctions and
matrix elements of the exchange interaction do not have singularities at the points of the degeneracy of two states as it takes place in the case of the parametric problem on the
semi‑axis. Our approach suggested allows the investigation of adiabatically driven quantum systems with a prescribed dependence on parametric adiabatic variables, as well. We
show that the choice of normalizing functions determining the transparent symmetric potential in fast variables leads to the zero coupling between states for two‑level systems,
(even at the point of
degeneration), while another choice of normalization factors leads to nonsymmetric potentials and nonzero coupling for the same energy levels.
New Binary Algorithm for the
Prediction of Protein Folding Types
Nikola Štambuk1, Paško Konjevoda1, and Nikola Gotovac2
1Ruđer Bošković Institute, Bijenička cesta 54, HR-10002 Zagreb, Croatia
2Department of Radiology, General Hospital
Požega, Osječka bb, HR-34000 Požega, Croatia
New binary algorithm for the prediction of α and β protein folding types from RNA, DNA and amino acid sequences is described. The algorithm was tested with machine
learning SMO classifier for the support vector machines and classification trees, on a dataset of 140 dissimilar protein folds. Depending on the method of testing, the overall
classification accuracy was > 90 and the tenfold cross-validation result of
the procedure was > 80%.
The method enables quick, simple and accurate prediction of α and β protein folds on a personal computer by means of few binary patterns of coded amino acid and
nucleotide physicochemical
properties.
Genetic code randomisation analysis based on 100,000 different codes tested for the protein fold prediction quality indicated that dipeptides represent basic protein units
with respect to the genetic code
defining of the secondary protein structure.
Molecular Modelling of Bioactive Gluten
Peptides and Related T-cell Receptors
Nikola Štambuk1, Duška Tješić-Drinković2, Ana Votava-Raić2, Paško Konjevoda1, Dorian Tješić-Drinković2, and Lana Mađerčić2
1Ruđer Bošković Institute, Bijenička cesta 54, HR-10002 Zagreb, Croatia
2Department of Pediatrics, Medical Faculty, University of Zagreb, Šalata 4, HR-10000 Zagreb, Croatia
Celiac disease is a common autoimmune condition caused by a selective lack of T cell tolerance for gluten. We investigated an algorithm for the prediction of T cell
stimulatory gluten peptides. The combined molecular modelling approach is based on the use of HLA‑DQ2 peptide binding motifs, the specificity of residues for tissue
transglutaminase, and the reconstruction/prediction of the interacting T-cell receptor sequence by means of the Molecular Recognition Theory. Modelled bioactive peptide
sequence was XQXPQXP and its predicted T-cell receptor target motif was heptapeptide PVHGVRG.
Statistical evidence for specific expansion of
the miRNA repertoire in vertebrates
Andrea Tanzer1,2 and Peter Stadler1,2
1Institute of Theoretical Chemistry and Structural
Biology, TBI, University of Vienna, A‑1090, Vienna, Austria
2Bioinformatics Group, Department of Computer Sciences &
Interdisciplinary Center of
Bioinformatics, University of Leipzig, D-04103 Leipzig, Germany
MicroRNAs (miRNAs) are a class of small non-coding RNAs found in most eukaryote genomes. Within the genome they reside either in introns or in non-coding regions as single genes or in
clusters of several different miRNAs within a range of a few kilo bases. MiRNAs are transcribed as long primary precursors (pri‑miRNAs), in the case of miRNA clusters as polycistronic RNA, of a
certain secondary structure containing several imperfect stem-loops. Maturation of miRNAs requires at least two compartmentalized processing steps: The nuclear RNAse III Drosha cuts off
these conserved stem-loops from pri-miRNAs resulting in precursor miRNAs (pre-miRNAs), also termed small temporal RNAs (stRNAs), of about 70-100 nt in length, which are exported to the
cytoplasm by means of the Exportin-5 pathway. In the cytoplasm Dicer, which is another protein complex with RNAse III activity, excises the mature miRNAs (~22 nt) from stRNAs.MicroRNAs seem to function in several different ways. The one best studied so far is translational retention. MiRNAs incorporated into RISC (RNA induced silencing complex) perform sequence
specific binding of RISC to target mRNAs in their 3' UTR (untranslated region) and by this inhibit termination of translation. During developmental processes it has been shown that miRNAs
inhibit expression of transcriptional repressors containing homeo domains which in turn initiates onset of further developmental steps.We recently reconstruct the evolution of the mir17 microRNA clusters. The history of this cluster is governed by an initial phase of local (tandem) duplications, a series of duplications of entire
clusters and subsequent loss of individual microRNAs from the resulting paralogous clusters. The complex history of the mir17 microRNA family appears to be closely linked to the early evolution
of the vertebrate lineage. Combining the evolution of coding as well as non-coding genes, we reconstructed the phylogenetic tree of two other miRNA, mir-10 and mir-196. In contrast to mir-17,
these genes apparently do not cluster with other miRNAs, but rather exists as single genes. The reason why we focused on them lies in their genomic context. Both reside within Hox gene
clusters, which consist of homeotic genes controlling body plan formation during early development. Further more, mir-196 has been shown experimentally to target three of the early Hox-genes.
Phylogenetic investigations of these miRNAs reflects the same evolution found for Hox genes. Like the members of the mir17 cluster, mir-196 seems to be vertebrate specific.
In contrast, mir‑10 is also found in non-vertebrates.Here we present statistical evidence that the expansion of the miRNA repertoire is linked to the origin of vertebrates. In general, major transitions in body plan require additional regulators, for
instance miRNAs. In vertebrates paralogos of several known miRNAs exist within each organism as described above. MiRNAs in non-vertebrates are found in vertebrates as well but not vice
versa. For instance Nematodes, Drosophila, Ciona and Echinodermata share more or less the same miRNAs, whereas Petromyzon, one of the basal vertebrates contains additional ones. Met-enkephalin effects on the histamine
induced bronchoconstriction in guinea-pigs
Dorian Tješić-Drinković1,
Nikola Štambuk2,
Duška Tješić-Drinković1,
Paško Konjevoda2,
and Biserka Pokrić2
1Department of Pediatrics,
Medical Faculty University of Zagreb, Šalata 4, HR-10000 Zagreb, Croatia
2Ruđer Bošković
Institute, Bijenička cesta 54, HR-10002 Zagreb, Croatia
We investigated the effects of neuropeptide met-enkephalin on the histamine induced bronhoconstriction in guinea-pigs. Konzett and Rössler’s method of whole body
pletismography modified by Gjuriš, was applied to investigate the effects. This method represents standard in vivo model of asthma. Dose-related modulatory effect of
met‑enkephalin on the bronchoconstrictor action of histamine was noticed and met‑enkephalin doses of 1 mg/kg and 10 mg/kg, respectively, caused significant reduction of
the histamine induced bronchoconstriction. Further studies are needed to define practical and therapeutical use of the presented observations in chronic obstructive
pulmonary diseases.
Redox state of residue Cys: which kind of information is stored in the
sequential vicinity
of these residues?
Éva Tüdős
and István Simon
Institute of Enzymology, Hungarian Academy of Sciences, H-1518, Budapest P.O.Box 7, Hungary
There are several methods to predict half cystine or free thiol cystein state of residue cys from the amino acid sequence of their flanking oligopeptides of various size. Due to
the high glutation concentration inside the cell practically all intracellular proteins have free thiol cysteins while most of the cys residues are in oxidized form, mainly half
cystine, in extracellular proteins. There is a significant difference in the amino acid composition of intracellular and extracellular proteins. In this lecture I will discuss the
question whether the sequence, or residue composition, in the vicinity of certain residues correlates with the potential of the cys residues to be in oxidized, mainly half cystine,
form or this sequence or residue composition identifies the extracellular or intracellular character of the protein, to which the cys residues belongs.
Course in Philosophy of Chemistry
Hrvoj Vančik
Department of Chemistry,Faculty of Science, University of Zagreb,
Strossmayerov trg 14, HR-10000 Zagreb, Croatia
It is an introductory course with the basic intention to represent the history of the Philosophy of Chemistry, and its establishment in the frame of philosophy of science. Note
that in the last ten years two scientific journals about this field have appeared. Besides such an introduction, some of main philosophical problems that are typical for
chemistry, as a scientific discipline will be discussed. To these topics also belongs discourse that includes emergence, supervenience, horizontal theories etc. Especial effort
will be done in the explanation of the theories about
complexity.
Binary substructure descriptors for organic
compounds
Kurt Varmuza, Wilhelm Demuth, and Heinz Scsibrany
Laboratory for Chemometrics, Vienna University of Technology, Austria
Among the many approaches for representing chemical structures as vectors, the method using binary substructure descriptors plays an important role. Substructures are
easily interpretable in terms of chemistry; they are capable to cover a great diversity of chemical structures, and substructure search is a standard operation in computer
chemistry.
Software SubMat was developed for easy and automatic calculations of binary substructure descriptors for a set of molecular structures and a set of substructures. SubMat
generates a text file with a line for each molecular structure that contains a string of 0´s and 1´s for absence or presence of the substructures. SubMat is running under MS
Windows; computing time for one molecular structure and 1000 substructures is typically 30 ms (Pentium IV, 2 GHz). SubMat can be optionally executed by calling it from
another program. In this case a command file is used to transfer file names and parameters to SubMat. During execution so called semaphore files are used to communicate
with the calling program and to
transfer error messages, status data, or a stop command.
A set of 1365 substructures has been defined covering a wide area of organic chemistry. A part of the substructures has been built systematically by using the isomer
generator software Molgen; others have been defined on the basis of chemical and spectroscopic ideas. The distribution of the substructures has been determined for two
spectroscopic databases.
Examples for using these substructures are from structure similarity searches, and evaluation of hitlists form spectra similarity searches. PCA and PLS are versatile tools for a
cluster analysis of chemical structures and spectra.
Acknowledgments to A. Kerber
and R. Laue (University of Bayreuth, Germany) for providing the isomer
generator software Molgen. Project P14792 of the Austrian Science Fund.
Quo vadis drug discovery?
Donatella Verbanac, Dubravko Jelić, Sanja Koštrun, Višnja Stepanić, and Dinko Žiher
PLIVA – Research Institute, Ltd., Prilaz baruna Filipovića 25,
HR-10000, Zagreb, Croatia
New century started with the characteristics of being landmark period for the science, medicine and drug discovery, as well. The drugs developed over the last four decades are
targeted at about 500 different biological targets. With the sequencing of the human genome, over 100,000 new biological targets will be recognized. It has been estimated that at
least 10 % of them could be used as targets for drugs. This will bring additional problems to be solved for an already “over- spread out” drug industry. There are a variety of
factors exerting increasing pressure on the discovery and development of novel drug candidates, not the least of these being the cost and time needed to pass through the various
phases of the process. Over the past
decade, a variety of scientific advances and economic pressures have driven the
need for improved drug discovery screening technology.
High throughput screening (HTS) is system for analyzing compound libraries and natural products in order to identify new therapeutic hits and leads on potential targets. In
combination with combinatorial chemistry it resulted in a paradigm shift from knowledge‑based sequential synthesis and testing to parallel processing of multiple compounds. With
the objective to improve success rates and cycle times for discovering new
hits, HTS is set to become one of the cornerstones of drug discovery.
In PLIVA - RESEARCH INSTITUTE Ltd. we introduced dedicated Screening Unit in mid 2000 with screenings of selected targets in anti-infective and anti-inflammatory area.
The compounds we screened are produced in house and are acquired from different providers. In response to the rapidly changing discovery paradigm, it is important to consider
preclinical properties at the earliest stages to assist in compound prioritization and to avoid downstream failure as related to drug delivery, pharmacokinetic or toxicological
performance. Implementing screens for physicochemical properties (e.g. ID/purity, stability, solubility, permeability) and in vitro metabolism can provide data to prioritize hits and
leads in favour of those most likely to possess acceptable preclinical properties. The design of these assays must appropriately balance throughput, accuracy and cost depending
upon the intended objective and at what phase of this discovery
process the data are needed.
In parallel, in silico data mining approaches such as high-throughput ADME/tox predictions and virtual screening including 2D and 3D methods are growing rapidly and are
ongoing in PLIVA - RESEARCH INSTITUTE Ltd.
An overview in developing an integrated approach in
providing these data, challenges for selected assays, and application to
hit-set deconvolution will be presented.
CHARACTERIZATION OF THE RESONANCE GRAPHS
OF CATACONDENSED HEXAGONAL GRAPHS
Aleksander
Vesel
Department of Mathematics and Computer
Science, PEF, University of Maribor, Koroška c. 160, SI-2000 Maribor, Slovenia
The vertex set of the resonance graph of a hexagonal graph G consists of 1‑factors of G, two 1‑factors being adjacent whenever their symmetric difference forms the edge
set of a hexagon of G.
A characterization of the resonance graphs of catacondensed hexagonal graph is presented. The characterization is the basis for the algorithm that recognizes the resonance
graph of a catacondensed hexagonal graph. Moreover, the modified algorithm can be applied
for recognizing the Fibonnacci cubes.
Valence Connectivity Versus Randić,
Zagreb and Modified Zagreb Index: A Linear Algorithm to Check Discriminative
Properties of Indices in Acyclic Molecular Graphs
Damir Vukičević1 and Ante Graovac2,3
1Department
of Mathematics, Faculty of Science, University of Split, Nikole Tesle 12, HR‑21000
Split, Croatia
2Department
of Chemistry, Faculty of Science, University of Split, Nikole Tesle 12, HR‑21000
Split, Croatia
3Ruđer
Bošković Institute, POB 180, HR-10002 Zagreb, Croatia
Valence connectivity in molecular graphs is described by 10-tuples μij where μij denotes the number of edges connecting vertices of valencies i and j. Shorter description is
offered by 4-tuples containing number
of vertices and values of Randić,
correspondence for all acyclic molecules of practical interest, i.e. for all of them having at most 100 atoms. This result is achieved by development of an efficient algorithm
which is linear in the number of 10-tuples.
Almost
all Trees and Chemical Trees Have Equiseparable Mates
Damir Vukičević1
and Ivan Gutman2
1Department of Mathematics, University of Split, HR-21000 Split, Croatia
2Faculty of Science, P. O. Box 60, 34000
Kragujevac, Serbia & Montenegro
Let T be an n-vertex
tree and e its edge. By ![]() and
and ![]() are denoted the number
of vertices of T lying on the two sides of e;
are denoted the number
of vertices of T lying on the two sides of e; ![]() .
.
Conventionally, ![]() . If
. If ![]() and
and ![]() are two trees
with the same number n of vertices, and if their edges
are two trees
with the same number n of vertices, and if their edges ![]() and
and ![]() can be
can be
labeled so that
![]() holds for all
holds for all ![]() , then
, then ![]() and
and ![]() are said to be equiseparable. Several previously studied
molecular–graph–based
are said to be equiseparable. Several previously studied
molecular–graph–based
structure–descriptors have equal values for equiseparable trees, which is a disadvantageous property of these descriptors. In earlier works large families of equiseparable
trees have been found. We now show that
equiseparability is ubiquitous and that almost all trees have an equiseparable
mate. The same is true for chemical trees.
Prediction of Consensus RNA Secondary
Structures Including Pseudoknots
Christina Witwer1, Ivo L. Hofacker1, and Peter F. Stadler1,2
1Institute for Theoretical Chemistry and
Structural Biology, University of Vienna, Währingerstraße 17/3, A-1090 Vienna,
Austria
2Bioinformatics Group, Department of Computer Sciences & Interdisciplinary Center of Bioinformatics,
University of
Leipzig, Kreuzstraße 7b, D-04103 Leipzig, Germany
Most functional RNA molecules have characteristic structures that are highly conserved in evolution. Many of them contain functional important pseudoknots. Here we
present a method for computing the consensus structures including pseudoknots based on alignments of a few sequences. The algorithm combines thermodynamic and
covariation information to assign scores to all possible base pairs, the base pairs are chosen with the help of the maximum weighted matching algorithm. We applied our
algorithm to a number of different types of RNA known to contain pseudoknots. All pseudoknots were predicted correctly, and more than 85% of the base pairs contained
in the reference structures were identified.
Thermoelectric properties of heavy fermions
Veljko Zlatić
Institute of Physics, HR-10002 Zagreb, Bijenička c. 46, Croatia
The temperature and pressure dependence of the thermoelectric power, S(T), and the electrical resistivity, ?(T), of heavy fermion intermetallic compounds, like CeRu2Ge2,
CeCu2Ge2 or CeCu2Si2 are explained by the single‑site Anderson model, which treats Ce‑ions as impurities but neglects coherent scattering on different Ce‑sites. The
pressure is taken into account by changing the hybridization between the f‑states and conduction band and we assume Ce3+ at ambient pressure. The ground state of such
a single‑ion model is always a Fermi liquid but at elevated temperatures we could have a magnetic or non‑magnetic fixed point, depending on the hybridization strength. The
properties of the model above the phase boundary line are calculated in the non‑crossing approximation, which shows that the seemingly complicated temperature
dependence of S(T) and ?(T), and their evolution as a function of pressure, is related to the crossovers between various fixed points of the model. Different pressure
dependencies of the energy scales
characterizing these fixed points account for the (T, p) phase diagram of the
above mentioned compounds.
On Modified Wiener Indices
Janez Žerovnik1,2
1Faculty of Mechanical Engineering, University of
Maribor, Smetanova 17, SI–2000 Maribor, Slovenia
2Department of Theoretical Computer Science,
IMFM, Jadranska 19, SI–1111 Ljubljana, Slovenia
The molecular–graph–based quantity W, introduced by Harold Wiener in 1947,8 nowadays known under the name Wiener number or Wiener index , is one of the most
studied molecular–structure–descriptors.5 A large number of modifications and extensions of the Wiener number was considered in the chemical literature; an extensive
bibliography on this matter can be found in the reviews.1 One of such modification, generalizing the idea of Nikolić, Trinajstić and Randić,4 was put forward by Gutman,
Vukičević and Žerovnik.3 The modified Wiener indices of a tree T are defined by mWλ(T) = ∑e [n1(e) · n2(e)] λ . It has been shown in the reference 3 that if trees are
ordered with regard to mWλ1 and mWλ2 for distinct λ1, λ2 < 0, then the two orderings are different. Analogous result was proved by Vukičević for λ1, λ2 > 0.6 In the proof
of the reference 3, the graphs used are not
necessarily of the same order, i.e. may have different numbers of vertices. In reference
2 it has been shown that
Theorem. For arbitrary different λ1, λ2 Î R there are graphs G1 and G2 of the same order such
that mWλ1 (G1) - mWλ1 (G2) < 0 and
mWλ2 (G1) - mWλ2 (G2) > 0 .
I will present
this result and some later related developments, most of them being joint work
with Damir Vukičević.7
1. M.V. Diudea, I. Gutman (1998)
Wiener-Type Topological Indices, Croat.
Chem. Acta 71: 21‑51.
2. M. Gorše, J. Žerovnik (2004) A remark
on modified Wiener indices, MATCH - Commun.
Math. Comput. Chem. 50: 109-116.
3. I. Gutman, D. Vukičević, J.
Žerovnik, A Class of Modified Wiener Indices, Croat. Chem. Acta, to appear.
4. S. Nikolić, N. Trinajstić,
M. Randić (2001) Wiener Index Revisited, Chem. Phys. Lett. 333:
319‑321.
5. N. Trinajstić (1992) Chemical Graph Theory, 2nd revised ed.,
CRC Press, Boca Raton.
6. D.
Vukičević (2003) Distinction Between Modifications of Wiener Indices,
MATCH - Commun. Math. Comput. Chem. 47: 87-105.
7. D. Vukičević, J. Žerovnik,
Variable Wiener Indices, MATCH - Commun.
Math. Comput. Chem., to appear.
8. H. Wiener (1947) Structural
Determination of Paraffin Boiling Points, J.
Am. Chem. Soc. 69: 17‑20.
Exact treatment of the interaction of an isolated state with the known
infinite‑dimensional quantum system
Tomislav P. Živković
Ruđer Bošković Institute, POB 180, HR-10002 Zagreb, Croatia
An
important problem in science is to describe the interaction of an isolated
state ![]() with the known
infinite dimensional quantum system
with the known
infinite dimensional quantum system ![]() . For example, one can consider
. For example, one can consider
the interaction of a molecule
with the electromagnetic field. To a very good approximation the interaction of
an isolated eigenstate ![]() of this molecule with
the electromagnetic
of this molecule with
the electromagnetic
field can be represented as the interaction of this eigenstate with the known infinite system (free electromagnetic field). Due to
this interaction, eigenvalue ![]() of
of ![]() is slightly
is slightly
shifted to
a new position. In addition, this eigenvalue is not sharp and it has the shape
of the universal resonance curve with finite uncertainty. If ![]() is an excited
molecular state,
is an excited
molecular state,
it is not stable and there is a finite probability for the transition of this state to other molecular states. All those properties are important experimental quantities. Investigation and
theoretical prediction of those properties is a main subject of spectroscopy. As another example consider the interaction of the molecule situated on the surface of some solid
with this
solid. One can again consider the interaction of the particular molecular eigenstate ![]() with this solid.
System
with this solid.
System ![]() represents a solid
with a surface. The solution to
represents a solid
with a surface. The solution to
this system usually consists of multiple eigenvalue bands. In addition, system ![]() may contain some
discrete eigenvalues that correspond to the surface states. Usually one
may contain some
discrete eigenvalues that correspond to the surface states. Usually one
knows
only an approximate solution of this system. Assuming this approximate solution
to be good enough, the problem is to find properties of the state ![]() subject to the
subject to the
interaction with this system. Investigation of this problem is one of the main
subjects of the surface state physics.
Standard treatment of this and similar problems is to use
perturbation expansion.1 However, if the interaction between the
state ![]() and the system
and the system ![]() is strong enough,
is strong enough,
perturbation expansion may diverge.1 Even if this expansion converges, this convergence can be unacceptably slow in order to obtain required accuracy. A new mathematical
approach for the exact quantum treatment of such systems was recently suggested.2,3 Unlike perturbation expansion approach this method produces exact quantum description
of the combined system, however
strong the interaction between the state ![]() and the system
and the system ![]() .
.
1.
A. Messiah (1965) Quantum Mechanics,
3rd ed., North-Holland, Amsterdam.
P.W. Atkins, R.S. Friedman (1997) Molecular Quantum Mechanics, 3rd ed., Oxford University
Press, Oxford.
2.
T.P. Živković (2002) J. Math. Chem.
32: 99-158.
3.
T.P. Živković (2004) Croat. Chem.
Acta, to be published.
Stationary processes in dilute gases and the maximum
entropy production principle
Paško Županović1, Edita Zenčić1, Davor Juretić1, and Srećko
Botrić2
1Faculty of Natural Sciences, Mathematics and
Education, University of Split, Split, Croatia
2Faculty of Electrical Engineering, Mechanical Engineering and Naval Architecture, University of Split, Split, Croatia
Transport Boltzmann equation is solved in the case of the stationary non‑equilibrium processes close to the equilibrium. It is shown that among all possible fluxes, which
ensure that the entropy produced in molecular collisions is equal to the entropy exported from the system, the actual fluxes are those that maximize the entropy production.