Abstract: MATH/CHEM/COMP 2005,
|
|
What can the computer say about the Randić
index? Mustapha Aouchiche1, Gilles Caporossi2, Pierre Hansen2 and Marie Laffay3 1Ecole 2GERAD and HEC 3CUST, In addition to performing numerous computational tasks in chemical graph theory, as e.g
computing chemical invariants or enumerating families of graphs, computers are being increasingly used for advancing the theory itself1, i.e.,
finding conjectures, proofs and refutations , in an assisted and sometimes fully automated way. The Randić index2 is one of the most studied among chemical invariants, both without and with computers. In this paper we survey computer-based work on the Randić index and its properties, and give some new results. We focus on: (i) finding and characterizing extremal graphs for the Randić index3; (ii) refuting, repairing, corroborating and strengthening Graffiti4 conjectures; (iii) proving automatically new conjectures and providing ideas of proof for more difficult ones with AGX5,6; (iv) making a
systematic comparison of the Randić index and other indices for various families of graphs, through conjecture finding with AGX. To complete the paper we prove, by computer or by hand, several conjectures, and present a
list of open ones. 1. P. Hansen (2002) Graph Theory Notes of 2. M. Randić (1975) J. Am. Chem. Soc. 97: 6609-6615. 3. G. Caporossi, 4. 5. G. Caporossi, P. Hansen (2000) Discrete Math. 212: 29-44. 6. G. Caporossi, P. Hansen (2004) Discrete Math. 276: 81-94. |
Abstract: MATH/CHEM/COMP 2005,
|
Fractal kinetics and its application to
BIACORE binding data analysis
Željko Bajzer, Yves Nominé
and Georges Mer Department of Biochemistry and Molecular Biology, The reaction environments
of in vivo conditions are characterized by spatial non-uniformity,
macromolecular crowding and small volumes1. In these situations it
is assumed that fractal geometry of the environment plays an essential role
and as a consequence the law of mass action valid for uniform environments
has to be modified. This leads to fractal–like kinetics1,2 which
implies time-dependent “rate constants”. We present a simple
derivation of law of mass action for reactions on fractals, based on
expression for the mean number of sites on the fractal visited by a random
walker in a given time period3. The model for bimolecular binding A + B
→ AB is developed and compared
to another model of fractal kinetics4 based on fractional powers
in classical low of mass action: k [A]α
[B]β In the recent years
measurement of binding of macromolecules have been performed by BIACORE. This
is an instrument where binding of molecules (A) in circulating solution to
molecules (B) attached to the surface of a sensor chip is recorded as a
function of time. The increase of mass on the chip due to binding is detected
by using surface plasmon resonance technology (www.biacore.com). It has been argued5,6 that
molecules attached on the chip surface constitute a fractal and that fractal
kinetics should be applied. Based on our equations for fractal bimolecular binding
we derive a model for binding in BIACORE instrument which also includes the
effects of flow. This model appears to be more realistic than the standard
model in which neither fractal surfaces, nor flow are taken into account. We
apply our model to reanalyze BIACORE data from literature7,8 and
we further analyze binding of breast-cancer-associated protein BRCA1 to phosphorylated DNA repair helicase BACH19,10.
1. S. Schnell, T.E. Turner (2004) Prog. Biophys. Molec. Biol. 85:
235-260. 2. R. Kopelman
(1988) Science 241: 1620-1626. 3. H.Q. Li, S.H.
Chen, H.M. Zhao (1990) Biophys. J. 58: 1313-1320. 4. M.A. Savageau
(2995) J. Theor. Biol. 176: 115-124. 5. A. Sadana (2001) Analy. Biochem. 291: 34-47. 6. Ž. Bajzer, J.D. Orth (20002) Biophys. J. 82: 481a. 7. R. Karlsson, A.
Michaelsson, Mattsson (1991) J. Immunol. Methods 145: 229-240. 8. H. Houshmand, G.
Fröman, G. Magnusson (1999) Anal. Biochem. 268:
363-370. 9. X. Yu, C.C.
Chini, M. He, G. Mer, J. Chen (2003) Science
302: 639-642. 10. M.V. Botuyan, Y. Nominé, X, Yu, N. Juranić, S.
Macura, J. Chen, G. Mer (2004) Structure
(Camb.) 12: 1137-1146. |
Abstract: MATH/CHEM/COMP 2005,
|
Heuristics for the Reversal Median Problem:
How to select good reversals?
Matthias Bernt, Daniel Merkle and Martin Middendorf Department of Computer
Science, The Reversal Median
Problem (RMP) is to find for three given signed permutations (genomes) a signed
permutation such that the number of reversals needed to transform it into the
given signed permutation is minimal. In Moret and
Siepel1 an exact branch-and-bound algorithm was
presented for RMP which can be applied in reasonable time only to small problem
instances. Other algorithms like MGR2 and rEvoluzer I3
use heuristic approaches that iteratively apply promising reversals starting
with the given signed permutations to find a solution. A new algorithm called
rEvoluzer II4 applies several promising reversals in parallel,
which leads to fronts of permutations approaching each other. Here we
investigate several strategies to select the candidate reversals for
obtaining the permutations in the front. Moreover, we study the properties of
the resulting solution sets for the RMP. 1. B. Moret, A. Siepel (2001)
Finding an optimal inversion median: Experimental results. In: Proc. of the 1st
International Workshop on Algorithms in Bioinformatics (WABI 2001),
No. 2149 in LNCS, Springer, pp. 189-203.
2. G. Bourque, P. Pevzner (2002)
Genome-scale evolution: Reconstructing gene orders in the ancestral species. Genome
Res. 12(1): 26-36. 3. M. Bernt, D. Merkle, M.
Middendorf (2004) Genome rearrangement based on reversals that preserve
conserved intervals. (submitted). 4. M. Bernt, D. Merkle, M. Middendorf (2005) A parallel
algorithm for solving the reversal median problem. (submitted). |
Abstract: MATH/CHEM/COMP 2005,
|
|
Yb3+
as an origin of the strong anti‑Stokes
luminescence in the NIR FT‑Raman spectra of some lanthanide sesquioxides Tomislav Biljan and Zlatko Meić Faculty of Science,
Department of Chemistry, Strossmayerov trg 14, 10000 Zagreb, Croatia Strong anti-Stokes bands
present in FT-Raman spectra of Y2O3, La2O3,
Gd2O3 and Lu2O3 are explained by
the NIR luminescence of Yb3+ impurities present in sesquioxides1
after the excitation with the 1064 nm line of a Nd:YAG laser. Samples of Y2O3:Yb,
La2O3:Yb, Ga2O3:Yb, CeO2:Yb,
Gd2O3:Yb and Lu2O3:Yb were prepared
by solution combustion synthesis procedure using urea. All materials were
investigated by FT-Raman and FT‑NIR spectroscopy and characterized by
X-ray powder diffraction. Strong anti-Stokes luminescence caused by Yb3+
ions in FT-Raman spectra has a potential application of qualitative and
quantitative determination of ytterbium in solid materials. 1. T. Biljan, S.
Rončević, Z. Meić, K. Kovač (2004) Chem. Phys. Lett.
395: 246–252. |
Abstract: MATH/CHEM/COMP 2005,
|
I-graphs and the Corresponding
Configurations
Marko Boben1,
Tomaž Pisanski2
and Arjana Žitnik3 1University of 2 University of 3
University
of Trivalent or cubic graphs
form an extensively studied class of graphs. Since they are sparse, trivalent
graphs can be readily drawn and visualized. Many graph theoretical problems
can be reduced to the trivalent case. The purpose of this presentation is the
study of I-graphs, a special class of trivalent graphs. I-graphs were
introduced in ref. 1 and form a natural generalization of generalized
Petersen graphs4. An I-graph is described by three integer
parameters. We determine the necessary and sufficient conditions for testing
whether two I‑graphs are isomorphic or not. We also classify I-graphs
in terms of girth, bipartiteness, and automorphism group. Bipartite cubic graphs
with girth at least 6 can be considered as incidence graphs (or Levi graphs)
of combinatorial configurations. Although configurations are mathematical
objects known for more than 150 years, the connection between them and
certain classes of graphs has not been widely investigated. But, for example,
the connection between configurations and cages was established in ref. 3.
Here we contribute some results concerning configurations arising from
I-graphs. From the combinatorial point of view,
some results follow from the properties of their Levi graphs, for example
about symmetry and about being triangle- or quadrangle-free, etc. From the
geometric point of view, there is an interesting connection to (cyclic)
astral configurations introduced in ref. 2. These configurations can be
realized in the Euclidean plane with maximal possible cyclic symmetry. 1. I.Z. Bouwer,
W.W. Chernoff, B. Monson, Z. Star (1988) The
Foster Census, Charles Babbage Research Centre. 2. B.
Grünbaum (1993) Astral (nk) configurations. Geombinatorics
3: 32–37. 3. T. Pisanski,
M. Boben, D. Marušič, A. Orbanić, A. Graovac (2004) The
10-cages and derived configurations. Discrete Math. 275: 265–276. 4. M. Watkins (1969)
A theorem on Tait colorings with an application to the generalized Petersen
graphs. J. Combin. Theory 6: 152–164. |
Abstract: MATH/CHEM/COMP 2005,
|
|
Mutability of short sequence repeats in human genome Branko Borštnik, Borut Oblak and Danilo Pumpernik National
The
availability of entire genomic sequences and many millions of single
nucleotide polymorphisms represents a challenge to computational chemists and
biologists. We have
combined both categories of information in order to unveil the susceptibility
of specific regions of human genome towards the alterations which lead to
genetic polymorphism. Since short sequence repeats are highly prone to
elongation and shortening process1,2 the attention was focused to
polyadenine repeats which we found several millions in human genomic
sequences and 55.000 among the single polymorphism entries. The polyadenines
were grouped in several categories and it was found that the short sequence
repeats in regions which are poor in genes represent the most mutable part of
human genome. 1. B. Borštnik, D. Pumpernik (2004) Europhys. Lett. 65:
290-296. 2. B. Borštnik, D. Pumpernik (2005) Phys. Rev. E 71: 031913(7 pages). |
Abstract: MATH/CHEM/COMP 2005,
|
MixeR package for compositional data analysis
Matevž Bren1,2 and Vladimir Batagelj 2,3 1Faculty of Organizational Sciences, 2Institute of Mathematics, Physics and Mechanics, 3Faculty of Mathematics and Physics, R
(http://www.r-project.org/) is `GNU S' - a language and environment for statistical
computing and graphics. R is similar to the award-winning S system, which was
developed at Bell Laboratories by John Chambers et al. It provides a wide
variety of statistical and graphical techniques (linear and nonlinear
modelling, statistical tests, time series analysis, classification,
clustering...). Further extensions can be provided as packages. We started to develop a
library of functions in R to support the analysis of mixtures and our goal is
a MixeR package for compositional data analysis that provides support for operations on compositions:
perturbation and power multiplication, subcomposition with or without
residuals, centering of the data, computing Aitchison's, Euclidean,
Bhattacharyya distances, compositional
Kullback-Leibler divergence etc. graphical presentation of compositions in ternary
diagrams and tetrahedrons with additional features: barycentre the geometric mean of
the data set, the
percentiles and ratio lines,
marking and coloring of subsets of the data set, notation of individual data
in the set etc. dealing with zeros and missing values in
compositional data sets with R procedures for simple and
multiplicative replacement strategy. We'll present the current
status of MixeR development and illustrate its use on selected data sets. J. Aitchison (1986) The Statistical Analysis of Compositional
Data, Chapman & Hall, New York. J.A. Martin-Fernandez, C. Barcelo-Vidal, V.
Pawlowsky-Glahn (2003) Dealing with zeros and missing values in compositional
data sets using nonparametric imputation. Math.
Geology 35 (3): 253-278. J.A. Martin-Fernandez, C. Barcelo-Vidal, M. Bren, V.
Pawlowsky-Glahn (1999) A measure of difference for compositional data based
on measures of divergence. In: Proceedings of the 5th Annual
Conference of the International Association for Mathematical Geology (S.J.
Lippard, A. Naess, R. Sinding-Larsen, eds.), Trondheim, Norway, vol. 1, pp. 211-215. H. Von Eynatten, C. Barcelo-Vidal, V. Pawlowsky-Glahn
(2003) Modelling compositional change: The example of chemical weathering of
granitoid rock. Math. Geology 35 (3): 231-251. |
Abstract: MATH/CHEM/COMP 2005,
|
Decomposition of the Free Energy Using
the Free Energy Perturbation Method
Urban Bren1 and Jan
Florian2 1Centre for Molecular
Modelling, National Institute of Chemistry, Hajdrihova 19, SI-1000 2Department of Chemistry, The partitioning of the free energy into
additive contributions originating from groups of atoms or force field terms
has a potential to provide free energy based relationships between structure
and biological activity of molecules and was used as a background for many
new methods of Bioinformatics like scoring functions or QSAR. Whether such
decomposition is justified has been a subject of vigorous debate in the
molecular modeling society. This question was never before addressed in terms
of the free energy perturbation (FEP) method, which represents the most
robust methodology for calculations of the free energy differences. Using the FEP methodology
we established, that every dissecting of the free energy into specific
components possesses an inherent error (termed the nonadditivity error)
arising from coupling between corresponding energy contributions. This effect
is seen as the appearance of mixing terms in the equation for the free energy
difference. Good news is that these mixing terms increasingly loose
importance as the change in the energy becomes smaller and smaller. Therefore
we are able to decrease the nonadditivity error to arbitrary value just by
increasing the number of FEP windows. At the same time one has to be aware,
that the free energy components are not state functions and should
consequently be calculated on the most natural trajectory possible. We confirmed our
theoretical findings via molecular dynamics calculations of hydration free
energies of nucleobases. Although our simulation was based on the
decomposition of the free energy, it yielded results surprisingly similar to
those obtained from the usual FEP procedure. Finally, we studied substituent
effects on 2’-deoxynucleosides with different charged substituents
attached at the 3’ position of deoxyribose. The obtained solvation free
energies of the base moieties exerted by their surroundings were only
negligibly affected by the structure and the charge of the substituent. The
nonadditivity error presented less than 0.1 % of the overall result. |
Abstract: MATH/CHEM/COMP 2005,
|
Studies of Homogeneous Electron Gas
Jerzy Cioslowski Emphasizing a proper
description of short-range interactions, the ladder theory (LT) is uncapable
of reliably reproducing any property of the three-dimensional electron gas
except for the correlation function at the electron coalescence limit (the
on-top density) g(0) and the related large-k tail of the momentum
distribution n(k). Because of the violation of the cusp condition, poor
accuracy of the predicted g(r) is expected for any nonvanishing r. Although LT yields components of the
correlation energy that satisfy the virial theorem for homogeneous interaction
potentials, in the case of the Coulomb potential these components turn out to
be infinite. A straightforward analysis shows that any effort at alleviating
this problem by introducing a long‑range screening is bound to violate
the virial condition. A commonly employed approximate version of LT, which
avoids Coulomb singularities, yields incorrect energy components and an
unphysical momentum distribution despite producing reasonable values of g(0).
Since lessening of the approximation worsens the accuracy of the high-density
limit of g(0), this result appears to be due to a fortuitous cancellation of
errors. |
Abstract: MATH/CHEM/COMP 2005,
|
|
On the use of ICS and CSA NMR data for predicting secondary structures of
peptides and proteins Attila G. Császár and Eszter Czinki Department
of Theoretical Chemistry, H-1117
Nuclear magnetic resonance
(NMR) chemical shielding tensors, computed at different levels of
Hartree-Fock, density functional (DFT), and even coupled-cluster [CCSD(T)]
theory within the gauge-including atomic orbital (GIAO) formalism, have been
investigated as a function of the backbone dihedral angles φ and ψ.
The changes in chemical
shieldings due to different residues and to the increase in model size are
investigated in detail. The computed surfaces are compared to results corresponding
to an experimental database. It appears that two
computational routes to structure determinations from NMR chemical shielding
information can be pursued. The perhaps simpler one concentrates on those
limited regions of the Ramachandran surface that characterize the dominant
peptide conformations. In this approach first all (or most of) the possible
conformers are obtained, and the NMR computations are performed at these
reference structures. This approach has been pursued, for example, for
β-hairpin conformers and dipeptide models involving diverse amino acid
residues. It seems that the most useful utilization of the corresponding
results is through the construction of multidimensional chemical shift/chemical shift plots. The second route does not
discriminate on the basis of conformers but defines complete isotropic
chemical shift (ICS) and chemical shielding anisotropy (CSA) surfaces for all
relevant nuclei as a function of dihedral angles. The periodic 3-dimensional
ICS(φ,ψ) and CSA(φ,ψ) surfaces, in the latter case
including both direction‑independent as well as directional
definitions, were fitted employing a number of suitable mathematical
functions. The best representation of the computed data is provided by a
10th-order cosine expansion. Although the CSA surfaces, when non‑directional
CSA definitions were used to generate them are well structured at the levels
of theory probed, the results suggest that these surfaces have only limited
utility in distinguishing between the major conformations, α-helix and
β-sheet, of peptides and most likely proteins. The orientation of NMR
chemical shielding tensors seems to depend substantially on the backbone
dihedral angles, most prominently in case of 13Cα,
and thus promises to provide useful information for distinguishing between
α-helix and β-sheet regions. For other nuclei, 1HN, 15NH,
13C, the orientation of the tensor is affected by other factors,
like H-bonding, and, consequently, the prediction of backbone angles using
tensor orientation information alone is limited. |
Abstract: MATH/CHEM/COMP 2005,
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Frequency analysis of photochemical
pollution data
Tomislav Cvitaš1,
Nenad Kezele1,
Leo Klasinc1, Matevž
Pompe2 and Marjan Veber2 1Ruđer Bošković Institute, Bijenička 54, 2Faculty of Chemistry and Chemical Technology, Frequency analysis of ozone data were recently reported for 12
European ozone monitoring sites1. The same procedure was applied
to the ozone and other pollutants data from six slovenian and croatian urban
stations. As expected, strong frequency signals are found for the 1 year and
1 day periods. The remaining peaks are selected statistically by checking whether the examined point
exceed the value of The most significant frequency peaks (Fig.1.) are
located at 365 and 1 day periods (natural cycles) and at 7 days period
(anthropogenic cycle). The relative intensity of 7 days peak could serve as a
indication of anthropogenic impact on measuring site. A quantification of the
degree of anthropogenic influences on ozone values was also given recently by
analysis of 7 days, 1 day and ½ day periodicities2. Some peaks in the spectra could correspond to exact
frequencies of the second, third and fourth harmonics of the base peak of
365.25 days. In most of the spectra, these peaks can be found on 6, 4 and 3
month periods. These harmonic cycles appear because the 1-year period is not
purely sinusoidal. Other important peaks for each station are found to
describe quasi periods ranging between 7 and 44 days and are listed in Table
1.
Table 1. Significant frequency
signals given in days. 1. T.
Cvitaš et al. (2004) J.
Geophys. Res. 109: 2302-2311. 2. N. Audiffren, C. Duroure,
G. Le Nir (2003) In: TOR-2 Final Report, EUROTRAC-2 ISS, GSF, |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Abstract: MATH/CHEM/COMP 2005,
|
|
Ab initio Studies on Low-Energy Conformers of
Oligopeptides Jiří Czernek Department of Bioanalogous and Special Polymers,
Institute of Macromolecular Chemistry, An accurate description of the
conformational space of peptides is of key interest in biomolecular science.
Thanks to the progress in the computer hardware and in the efficiency of the
relevant software, ab initio quantum chemical methods can now
routinely be applied to quite sizeable peptide structures (hundreds of
atoms). In particular, the resolution-of-identity (RI) approximation
elaborated by Ahlrichs and coworkers and implemented in the TURBOMOLE program
package enables one, when combined with some density functional theory
(DFT)-based method, to perform computationally very efficient geometrical
optimizations. Moreover, RI approximation coupled with the second-order Møller–Plesset
perturbational theory (MP2) can account for a large portion of the
correlation energy with reasonable computer time, memory, and disc space
requirements. A number of methods, both RI-based and conventional, have been
applied to describe the geometries and total energies of four low-energy
conformers of an important model system, N‑acetyl‑N’‑methylalanineamide
(“dialanine”), which has been studied at the Hartree–Fock
level previously1. The basis set dependence of the results has
been addressed by performing each calculation with a smaller (i.e., SVP) and
an extended (i.e., TZVP) set of atomic orbitals. The accuracy of the relative
energies provided by respective approaches has been assessed by a comparison
with the benchmark RI-CC2 (the second order approximate coupled cluster
method)/TZVP results. Seven conformers of the heptapeptide Ac‑VVVE(Hnb)(tBu)VVV‑OH
(Ac, V, and E denote acetyl group, valine, and glutamic acid, respectively,
Hnb is 2-hydroxy-6-nitrobenzyl group used to substitute the amide proton of
E, and tBu is tert-butyl group protecting the carboxylic function of E),
which consists of 148 atoms and has been prepared in our Department and
studied experimentally and by molecular dynamics, have been subsequently
optimized by RI‑DFT methods and their final energies have been calculated
using the RI-MP2 technique. Various factors contributing to each
conformer’s stability are compared. The importance of the inclusion of
the correlation energy in the calculations is highlighted. Acknowledgements. This
research has been supported by the 1. H.-J. Böhm, S. Brode (1991) J. Am. Chem. Soc. 113: 7129–7135. |
Abstract: MATH/CHEM/COMP 2005,
|
Testing of a priori vapor pressure and
boiling point predictive ability of COSMOtherm
András
Dallos and R. Kresz The COSMOtherm program is based on COSMO-RS
theory of interacting molecular surface charges1. The chemical
potential of the compounds in the system, which allows for the prediction of
almost all thermodynamic properties of compounds or mixtures, is available
from integration of the screening charge density over the surface of the
compounds2. In addition to the prediction of thermodynamics of
liquids COSMO-RS is also able to provide a reasonable estimate of a pure compound’s
chemical potential in the gas phase and therefore it is possible to a priori predict vapor pressures of
pure compounds. For testing of vapor pressure and boiling point
predictive ability of COSMOtherm we compared the experimental vapor
pressure values at 298.15 K and normal boiling points of 139 organic
compounds of 20 molecular classes, mostly homologues (normal-, iso-, cyclo-,
1-chloro-, 1-bromo-, 1-fluoro-, 1-cyano-, 1‑nitro-, 1-acetoxy-alkanes,
alkenes, alkynes, aromatics, 1-alkanols, 2-alkanols, 1-thiols, 2-alkanones,
aldehydes, esters, pyridins and others) with those estimated by COSMOtherm.
It was concluded that the estimation power of COSMOtherm is close to
the vapor pressure calculation methods with one adjustable parameter, like
Thek-Stiel extrapolation method.  (a) (b) Figure 1. Comparison of measured and estimated vapor pressure of benzyl acetate
at different temperatures (a) and 139 organic compounds at 298.15 K (b). 1. A. Klamt
(1995) J. Phys. Chem. 99: 2224-2235. 2. A. Klamt, F. Eckert (2000) Fluid Phase Equilibr. 172: 43-72. |
Abstract: MATH/CHEM/COMP 2005,
|
Chemical Ontologies
for Bioinformatics
Kirill Degtyarenko
European Bioinformatics Institute, Wellcome Trust
Genome Campus, Hinxton, Although biological macromolecules (nucleic
acids and proteins) are biochemical entities, the biological sequence
databases often lack meaningful (bio)chemical annotation. To process the
ever-growing volume of biological sequence data, the standardization and
organization on different levels is required, from controlled vocabularies to
dictionaries and thesauri to taxonomies and formal ontologies. Ontology of
some domain of knowledge is defined here as a controlled vocabulary of terms
with defined logical relationships to each other. The unique types of
relationships between terms have to be included in chemical ontologies. To
illustrate the relevance of chemical ontologies to bioinformatics, the
current resources at the European Bioinformatics Institute are presented,
such as ChEBI (Chemical Entities of Biological Interest), COMe (the ontology
of bioinorganic proteins) and the ontologies for physico-chemical methods and
processes. |
Abstract: MATH/CHEM/COMP 2005,
|
|
Special properties of density matrices derived from an arbitrary full-CI wave function Gergey Dezsö1,2 and Iván Gyémánt1 1Department of Theoretical Physics, 2Department of Technology
and Production Engineering, The importance of density
matrices in physics and chemistry is known since the dawn of quantum
mechanics. The main advantage of describing a system involving at most two‑particle
interactions by density matrices, particularly second-order reduced density
matrices is, that it contains all relevant information in the most compact
form. Unfortunately, second-order reduced density matrices can not be
optimized directly because of the so-called N-representability problem. Since the
N-representability problem is realized, there were a lot of attempts to
formulate necessary and sufficient conditions for density matrices ensuring
their N-representability without the knowledge of the underlying wave
function. Nowadays we have either necessary or sufficient conditions,
excellent approximate methods, but the exact solution of the
N-representability problem is not known. Our approach works within
the frame of a finite one-particle basis, but the dimension of this basis is
arbitrary. Density matrices can be expanded in geminal basis, where geminals
are antisymmetric two-particle functions constructed over the one-particle
basis. N-representable density matrices must be derivable from a FCI-type wave
function, which is a linear combination of Slater-determinants over the
one-particle basis. In previous papers the connection between the wave
function and the density matrix was exactly described by the so-called
structure matrix1. This enables one to optimize the density
matrice2-4, but configurational coefficients are needed in each
iteration step, when building the density matrix and the gradients of the
matrix elements. Elements of the density
matrix (in the geminal basis) are pointed out to be written exactly as scalar
products of specially defined vectors. These vectors contain the
configurational coefficients. Using basic rules of vector operations,
conditions can be formulated including only elements of density matrix.
Necessary conditions formulated have the form of inequalities. Case studies for small atoms and
molecules are also presented. 1. I.
Bálint, G. Dezsö, 2. I.
Bálint, G. Dezsö, I. Gyémánt (2001) J. Chem. Inf. Comp. Sci. 41: 806-810. 3. G. Dezsö,
I. Bálint, 4. I.
Bálint, G. Dezsö, |
Abstract: MATH/CHEM/COMP 2005,
|
||||||
|
Generalized Operations on Maps Mircea V. Diudea1, Monica Ştefu1, Peter E. John2 and Ante Graovac3,4 1Faculty of Chemistry and Chemical Engineering, “Babeş-Bolyai” University, 400028 3 Ruđer Bošković Institute, HR-10002 4Faculty of
Natural Science, Mathematics and Education, University of Split, N.
Tesle 12, HR-21000 Split, Croatia A map M is a combinatorial representation of a closed surface. Convex
polyhedra, starting from the Platonic solids and going to spherical
fullerenes, can be operated to obtain new objects, with a larger number of
vertices and various tiling. Three composite map
operations: leapfrog, chamfering and capra, play a central role in the
fullerenes construction and their electronic properties. Generalization of the
above operations leads to series of transformations, characterized by
distinct, successive pairs in the Goldberg multiplication formula m(a,b) see Figure 1. Parents and products of most representative operations are illustrated.
|
||||||


Abstract: MATH/CHEM/COMP 2005,
|
|
Transcription Factor Target Detection in Comparative Genomics Claudia Fried and Peter F. Stadler Bioinformatics
Group, Department of Computer Science, University of Leipzig,
Hätelstraβe 16-18, D-04107 Leipzig, Germany Cellular signaling
pathways induce gene expression by activating specific transcription factors.
Errors in the activation of transcription factors are common events in cancer
as several of this factors act on genes involved in ellular proliferation,
survival and differentiation. The identification of the targets of tumorigenic transcription factors that
cause these changes is an important task in cancer research. One way to identify
those target genes is detection of transcription factor binding sites. These
binding sites can be predicted by the search of recurring motifs in the
regulatory regions of co-expressed genes1. This has been shown to
be feasible in yeast where the intergenic regions are very small. On the
other hand, intergenic regions in the genomes of vertebrates can be very
large. A simple search for exact string matches of experimentally verified
binding sides on a genome wide level in vertebrates thus leads to a high
number of false positives. To overcome this problem, only regions might be
taken into account that are evolutionarily conserved. Conserved regions can
be detected by phylogenetic footprinting with the program tracker2
that compares non-coding sequences surrounding a set of orthologous genes.
The aim of our study, is to find target genes of the transcription factor
STAT3 (Signal transducer and
activator of transcription 3), a member of the STAT family of transcription
factors that act as signal transducers of cytokines, hormones and growth
factors. Stat3 is involved in the regulation of cell growth, survival and
differentiation3. Analysis of STAT3 targets found by this study
can provide new insight into mechanisms of cancer and may shed light on
strategies for targeted therapy. 1. C. Dieterich, R. Herwig, M. Vingron
(2003) Bioinformatics 19. 2. S. Prohaska, C. Fried, C. Flamm, G. P. Wagner, P.F.
Stadler (2004) Mol. Evol.
Phylog. 31: 581-604. 3. J. Turkson
(2004) Expert. Opin. Ther. Targets.
8 (5): 409-422. |
Abstract: MATH/CHEM/COMP 2005,
|
|
QSPR
STUDY OF FLAVONOID COMPOUNDS Josipa Friščić1, Sonja Nikolić1 and Marica Medić-Šarić2 1Ruđer Bošković Institute, HR-10002 2Faculty of Pharmacy and Biochemistry, Flavonoids are a group
of coumpounds widely distributed in plants, and due to their poliphenolic
structure on flavan nucleus (Fig.1.) they have significant antioxidant and
chelating properties.
Figure 1. Flavan nucleus. Their antibacterial, antiviral, antimycotic,
antiinflammatory and antithrombotic effects have been proven, as well as
their positive effects on tumors, cardiovascular, immunological and many
other diseases. Structure diversity and various mechanisms of actions, as
well as great number of methods for establishing their activity, makes
flavonoids a challenge for making a complete QSPR/QSAR profile1. In this work, several QSPR (Quantitative Structure-Property Relationship) models in study of
some physical‑chemical properties (partition coefficient logP, Van der
Waals volume Vw, molecular weight, and melting point) have been
investigated for group of 30 flavonoids. For calculating the molecular
descriptors we used 2 computer programs – TAM2 (University
of Zagreb) and DRAGON 3.03 (University of Milan). Models were
calculated using program CROMRsel4 (Ruđer Bošković
Insitute in Zagreb). All models were tested for their stability and
predictability, and we compared models obtained with molecular descriptors
from TAM and from DRAGON 3.0. It has been shown that for simple models we can use molecular descriptors calculated with TAM (molecular weight), while molecular descriptors from DRAGON 3.0 give more reliable models. For predicting melting point no reliable models were obtained. For partition coefficient, log P, and Van der Waals volume, Vw, best models were those with 2 molecular descriptors calculated with DRAGON 3.0 (Fig. 2.).
1. K.E. Heim, A.R. Tagliaferro, D.J. Bobilya
(2002) J. Nutr. Biochem. 13: 572-584. 2. S.
Marković, M. Vedrina, M. Medić-Šarić, N. Trinajstić
(1997) Comput. Chem., 21: 355-361. 3. R. Todeschini, V. Consonni (2000) Handbook of Molecular Descriptors,
Wiley-VCH, Weiheim. 4. B. Lučić, N. Trinajstić (1999) J. Chem. Inf. Comput. Sci. 39: 121-122. |



Abstract: MATH/CHEM/COMP 2005,
|
Partial Orders, Molecular Phylogenies,
and Migration
Guido Fritzsch1,2,
Sonja J. Prohaska 1,3
and Peter F. Stadler1,3,4,5 1Interdisciplinary 2Molecular Evolution and Animal Systematics,
Departmen of Biology II, 3Bioinformatics, Department of Computer Science, 4Institute for Theoretical Chemisty, 5The Santa Fe Institute, The
expansion of a species in a heterogeneous environment can be correlated with
relative rates of evolution in geographically separated subpopulations. The
rate variation may be due to adaptation to different environmental conditions
and due to changes in population size or structure1. Given
a phylogenetic tree, Tajima's relative rate test2 can be used to
identify pairs of taxa that have evolved with significantly different rates
since their divergence from a common ancestor. The ratio of the inferred
number of substitutions since this last common ancestor can then be used as
an estimate for the ratio of
their speed of evolution. We use the quantity exy
which is defined as the average of the logarithms of the ratios over all
possible choices of the outgroup using the chi-squared value of the Tajima
tests as weights. One
can show that the matrix (exy)
is anti-symmetric and transitive, i.e., exy >0 and
eyz >0 implies exz >0. Thus it defines
a partial order on the set of taxa that faithfully reflects the relatives
speed of evolution:
Figure 1. Example
of a relative rate poset. Data are 5'UTRs of HIV-1. Thin lines in the l.h.s.
panel indicate significant Tajima tests, the thick lines represent the
associated Hasse diagram of the
partially ordered set.
We analyse the relative rate posets of a variety of cases
in which recent migrations are well documented and for which a fairly
complete taxon sampling is available, among them the European pond turtle Emys
orbicularis3. Acknowledgements. This work was supported in part by the DFG
Bioinformatics Initiative. 1. C. Stringer,
R. McKie (1996) African Exodus: The Origins of Modern Humanity. John MacRae Book/ 2. F. Tajima (1993) Genetics 135: 599-607. 3. P. Lenk, U. Fritz, U. Joger, M. Wink (1999) Mol. Ecol. 8:1911-1922. |

Abstract: MATH/CHEM/COMP 2005,
|
||||||||
|
Sphericities of Cycles. What Pólya’s Theorem is Deficient in Shinsaku Fujita Department of Chemistry and Materials Technology,
Kyoto Institute of Technology, Matsugasaki, Sakyoku, Kyoto, 606-8585 Japan 1. Introduction The concepts of sphericity, sphericity
indices, and unit subduced cycleindices with chirality fittingness
(USCI-CFs), which have been proposed by Fujita on the basis of coset representations
and their subductions1, are versatile to discuss stereochemistry
in molecule as well as to enumeratestereoisomers.This USCI approach is
capable of enumerating isomers as 3D chemical structures (stereoisomers),
where theyare itemized with respect to point-group symmetries. However, the USCI
approach requires mark tables, USCI tables, and related group theoretical
tools, which are not so easily obtained. On the other hand, Pólya’s Theorem, which has been widely
used from 1930s2, is simple and convenient to calculate gross
isomer numbers without taking account of symmetry-itemization. Because Pólya’s Theorem enumerates graphs, not 3D
chemical structures (stereoisomers) from the viewpoint of the USCI approach,
its application has been restricted within graph-theoretical problems. In
order to treat such stereochemical problems as solved bythe USCI approach,
what is Pólya’s Theorem deficient in? 2. The Proligand Method The deficiency of Pólya’s Theorem is now concluded to be
the concept of sphericities of cycles3. For example, let us
consider an enumeration problem in which the three hydrogen atoms of acetic
acid are replaced by Fand p (andp), where chiral proligands pandpare
enantiomeric to each other. After the three hydrogens are numbered
sequentially, the relationship between each symmetry operation and the
resulting permutation (a product of cycles) is examined to give the following
correspondence:
In this table, a permutation corresponding to an
improper rotation is called an improper pemutation and designated by an
overbar. The cycles are divided into three categories and characterized
by sphericities and sphericityindices. Thus, an odd-membered
cycle contained in an improper permutation is called a homospheric cycle and
characterized by a sphericity index ad (d: the length of the
cycle), while an even-membered cycle contained in an improper permutation
is called an enantiospheric cycle and characterized by a sphericity
index cd. On the other hand, a cycle contained in a proper
permutation is called a hemispheric cycle and characterized by a
sphericity index bd whether d is odd or
even.Thereby, the permutations listed above are characterized by products of
such sphericity indices, which are summed up to give a CI-CF (cycle
index with chirality fittingness):
The
CI-CF provides us with a tool (the proligand method) for stereoisomer
enumeration, in which the chirality fittingness controls the mode of
accommodation ofchiral and achiral ligands. By placing ad = cd = bd = sd, we can obtain the cycle index of
Pólya’s Theorem (i.e., CI =
(1/6)(s13+2s3+3s1s2)). This means that the
proligand method is more informative than Pólya’s theorem so as
to be capable of solving stereochemical problems. 1. S. Fujita
(1991) Symmetry and Combinatorial Enumeration
in Chemistry, Springer-Verlag. 2. G.
Pólya, R. C. Read (1987) Combinatorial
Enumeration of Groups, Graphs, and Chemical Compounds, Springer-Verlag. 3. |
Abstract: MATH/CHEM/COMP 2005,
|
Solid-State
and Solution-State Conformational Differences Solution-State and 13C CP/MAS NMR Studies on Conformational
Polymorphic
|
Abstract: MATH/CHEM/COMP 2005,
|
The Evolution of Animal
miRNAs
Jana
Hertel1,
Claudia Fried1,
Manja Lindemeyer1,
Kristin Missal1, Sonja J. Prohaska1, Andrea Tanzer1,2, Christoph Flamm2, Ivo L. Hofacker2, Peter F. Stadler1,2,3, and the
students of the bioinformatics computer labs 2004 and 2005 1Bioinformatics Group, Department of
Computer Science, 1
2 Institute for Theoretical Chemistry and
Structural Biology, 2
3
3The
Santa Fe Institute, MicroRNAs are short
(~22nt) non-coding RNAs, that act as factors in the degeneration or translational
repression of their mRNA targets1. We clustered the known miRNAs
due to their sequence similarity and searched all available metazoan genomes
for homologs. Based upon those hits we constructed gene phylogenies. All of
the resulting trees are consistent with the genome duplication history at the
origin of the vertebrates and the teleost lineage, respectively. Tandem
duplications of microRNAs typically preceded these genome-wide events. We furthermore
investigated distant homologies between established microRNA families. To do
that we aligned their consensus sequences and examine the resulting phylogeny
for the known phylogenetic theories. The intention to this is to identify
larger groups that could be distant homologs. Recently, genome-wide surveys
have identified thousands of putative non-coding RNAs2. Our
exhaustive analyses provide a phylogenetic grouping of microRNAs that serves
a basis for the development of computational methods for microRNA
recoginition. This will lead us to the classification of microRNAs within a
large collection of non-coding RNAs. Acknowledgements. This work was supported in part by
the DFG Bioinformatics Initiative. 1.
P. Nelson, M. Kiriakidou, A. Sharma, 2. S. Washietl, I.L. Hofacker, P.F. Stadler (2005) Thousands of noncoding
RNAs with conserved structure in mammalian genomes. (in review) |
Abstract: MATH/CHEM/COMP 2005,
|
The Redundancy of Topological Indices
Boris Hollas Theoretische
Informatik, Universität Ulm, D-89069 Ulm One approach to predict or
compare molecular properties is to encode a molecule by a set of numbers and
to use these molecular descriptors for
regression analysis or clustering. Topological
indices (TIs) are molecular descriptors based on the graph of a molecule.
A large number of TIs has been proposed and applied in QSAR and QSPR studies,
a method to relate the structur of a molecule to a property (e.g. boiling
point or toxicity). Many TIs are mutually correlated1, which causes major problems
in QSAR and QSPR studies. Most QSAR/QSPR approaches use molecular descriptors
to establish a linear regression model. If descriptors are correlated, the
outcome of this model becomes meaningless or modeling may fail completely. In a series of papers2-6, we consider TIs of the
general form
where
It
turns out that TIs Another
issue is the variance of 1.
I. Motoc, A.T. Balaban, O. Mekenyan, D. Bonchev (1982) MATCH Commun. Math. Comput. Chem. 13: 369-404. 2.
B. Hollas (2002) MATCH Commun. Math. Comput. Chem. 45: 27-33. 3.
B. Hollas (2003) J. Math. Chem. 33: 91-101. 4.
B. Hollas (2003) MATCH Commun. Math. Comput. Chem. 47: 79-86. 5.
B. Hollas (2005) MATCH Commun. Math. Comput. Chem. 54: 177-187. 6.
B. Hollas (2005) MATCH Commun. Math. Comput. Chem. 55 (to appear) |
Abstract: MATH/CHEM/COMP 2005,
|
On the Number of
Hamiltonian Groups
Boris Horvat,
Gašper Jaklič and Tomaž Pisanski
IMFM,
Jadranska 19, SI-1000 Subgroups of abelian
groups are abelian and hence self-conjugate or normal. A nonabelian group all
of whose subgroups are normal is called hamiltonian1. In topological graph
theory2,5, hamiltonian groups have been studied in the past3,4.
For several classes of hamiltonian groups the genus is known exactly. For
abelian and hamiltonian groups, there are structural theorems available. In this paper we determine
the number h(n) of hamiltonian
groups of order n and the number b(n) of all groups of order n with the property, that all their
subgroups are normal. We also determine the number v(n) of all hamiltonian groups of order ≤ n and the number w(n) of all groups of
order ≤ n with the property,
that all their subgroups are normal. 1. R.D. Carmichael (1956) Introduction to the Theory of Groups of
Finite Order, Dover Publishing Co., 2. J.L. Gross, T.W. Tucker
(1987) Topological Graph Theory,
Wiley 3. T. Pisanski, T.W. Tucker
(1989) The genus of the product of a group with an abelian group. Eur. J. Combin.10: 469‑475. 4. T. Pisanski, T.W.
Tucker (1989) The genus of low rank hamiltonian groups. Discrete Math. 78:
157-167. 5. A.T. White (2001) Graphs of Groups on Surfaces,
North-Holland Publishing Co., |
Abstract: MATH/CHEM/COMP 2005,
|
The SOS response signalling mechanism in bacteria Escherichia coli: Involvement of RecA loading activity
Ivana Ivančić-Baće1,
Ignacija Vlašić2,
Boris Mihaljević2,
Mirna Imešek2, Erika Salaj-Šmic2 and Krunoslav Brčić-Kostić2 Department of Molecular Biology,
Faculty of Science, Rooseveltov trg 6,
Department of Molecular Biology, Ruđer
Bošković Institute, Bijenička 54, The SOS response involves many bacterial functions that are induced in response to damage of chromosomal DNA. The SOS response is regulated by the LexA repressor and the RecA nucleofilament which is formed at single stranded DNA. Such RecA nucleofilaments display a coprotease activity that stimulates self-cleavage of the LexA repressor. This cleavage enables transcription of more that 40 genes in the SOS pathway. The exact mechanism whereby initial
damage leads to induction of SOS response is still unknown. It has been
proposed that the signal for SOS induction is single-stranded (ss) DNA formed
as a result of DNA damage. This ssDNA can be produced either by DNA
polymerase arresting at sites of DNA damage or by RecBCD enzyme unwinding at
dsDNA breaks. RecA protein then probably forms presynaptic filaments on these
regions of ssDNA, resulting in repressor cleavage. We wanted to test our
hypothesis that RecA loading is essential step in creating the SOS inducing
signal. In order to test this, we measured SOS response in a strain in which
the lacZ gene (expressing b-galactosidase) is
fused with the regulatory region of a sfiA SOS gene. We introduced
additional mutations into this strain that inhibit RecA loading and nuclease activity
but retain helicase activity (recB1080 and recB1067 mutations).
We also wanted to test whether different types of DNA damage require
different enzyme processing. Therefore we measured SOS induction after UV and
gamma irradiation, and introduction of double strand breaks by endonuclease.
Bacteria were grown at 30°C in
LB broth. When cultures reached O.D.600=0.2, cells were either UV‑irradiated,
g-irradiated
or arabinose was added at final conc. 0.2%. Addition of arabinose induces
SceI endonuclease which introduces dsDNA breaks in DNA. The cultures were
further incubated for 180 minutes and assayed for β-galactosidase
activity. Our results indicate that
RecA loading activity is required for SOS inducing signal after UV,
γ-irradiation and dsDNA breaks. When RecA loading activity is inactivated
in RecB1080CD enzyme, this activity is provided by RecFOR. |
Abstract: MATH/CHEM/COMP 2005,
|
Analytical treatment of hydrogen vibrations
in molecular dynamics simulation
Dušanka Janežič, Franci Merzel and
Matej Praprotnik Laboratory
for Molecular Modeling and NMR, National Institute of Chemistry, We propose an analytical
treatment of hydrogen bond-stretching vibrations in molecular dynamics
simulations using a new form of the classical Liouville propagator. We
construct a second-order integrating algorithm which is useful for all-atom
molecular dynamics simulations of molecular systems described by flexible
models. We apply this algorithm to MD simulation of the liquid water
providing the evidence about its superior numerical characteristics over the
standard approach. |
Abstract: MATH/CHEM/COMP 2005,
|
A Multi-Objective Particle Swarm Optimization
|

Abstract: MATH/CHEM/COMP 2005,
|
A 2-parametric class of transformations for
fullerenes and other polyhedra
Peter E. John and Horst Sachs Let
P denote any polyhedron all of whose vertices have valency 3, and let S
denote the set of all such polyhedra. A 2-parametric class of transformations
T, generalizing the well-known leapfrog procedure and related operations, is
defined which maps S into S and, in a way, preserves the relative position of
the non‑hexagonal faces of P. Every
such transformation T – except for two “small” cases
– has the property that each of the polyhedra T(P) possesses some
perfect matching that avoids any edge that belongs to the boundary of some
non‑hexagon. |
Abstract: MATH/CHEM/COMP 2005,
|
|
De Novo Determination of Protein Substructures from NMR Couplings of Peptide Groups Nenad Juranić, Franklyn
G. Prendergast and
Slobodan Macura Departments of
Biochemistry and Molecular Biology , Mayo College of Medicine, Mayo Clinic
and Foundation, Rochester, Minnesota, 55905 U.S.A Multi-dimensional NMR spectroscopy
has been an effective tool for determining three-dimensional structures of
relatively small proteins (<25 kD). Its application to larger proteins
faces several challenges the most serious of which is that the fast
relaxation of nuclear spins in such samples makes deuteration (proton-depletion) an
absolute requirement1. While proton-depletion enables the use of
heteronuclear correlation experiments to determine backbone assignments, it
severely reduces the structural information obtained regarding interproton
distances, traditionally the major source of structural constraints2.
However, new structural constraints, that can be obtained from residual
dipolar couplings3-5 and hydrogen-bond spin-spin couplings6-8,
have been introduced, and these offer the possibility for atomic-resolution
structure determination of the
protein backbone. In principle,
protein structures can be solved from residual dipolar couplings alone, but
that requires multiple orientation media (needs five independent alignments)9
which is impractical and often impossible because of the adverse
protein-media interactions. Consequently, methods that rely on trial
substructures are currently used. A more objective approach requires the
experimental determination of an initial substructure and a protocol for
direct structure building. We have developed method to solve an initial substructure by utilizing sequential and hydrogen-bond connectivity of peptide groups (as detected by NMR spin-spin couplings). The initial substructure has such accuracy that it allows determination of protein alignment tensor using a single orientation media. Once the alignment tensor is known, we propose direct protein structure building utilizing NMR detected hydrogen bond chains8, which are essential elements of protein secondary structure.
Figure 1: Utilizing the ideal hydrogen bond chain motifs (HBCs), a model of the initial sub-structure was obtained
from three neighboring HBCs containing high values of the H-bond couplings,
while the connectivity between them required an anti-parallel type of HBC
1. R. A.Venters, R. Thompson, J. Cavanagh
(2002) J. Mol. Struct. 602: 275. 2. S. Macura, R. R. Ernst (2002)
(reprinted from Mol. Physics (1980)
41: 95-117), Mol.Physics 100: 135. 3. J. R. Tolman, J. M.
Flanagan, M. A. Kennedy, J. H. Prestegard (1995) P. Natl. Acad. Sci.USA 92:
9279. 4. N. Tjandra, A. Bax (1997) Science 278: 1111. 5. A. Bax, G. Kontaxis,
N. Tjandra (2001) Pt B. 339: 127. 6. E. Cordier, S.
Grzesiek (1999) J. Am. Chem.
Soc. 121:
1601. 7. G. Cornilescu, J. S. Hu, A. Bax (1999) J. Am. Chem. Soc. 121: 2949. 8. N. Juranic, M. C. Moncrieffe, V. A.
Likic, F. G. Prendergast, S. Macura (2002) J. Am. Chem. Soc. 124:
14221. 9.
K. B. Briggman, J. R. Tolman (2003) J. Am. Chem. Soc. 125: 10164. |

Abstract: MATH/CHEM/COMP 2005,
|
|
The maximum entropy production principle as the guideline for predicting evolution of complex systems Davor Juretić1, Paško Županović1 and Srećko Botrić2 1Faculty
of Natural Sciences, Mathematics and Education, University of Split 2Faculty of Electrical Engineering,
Mechanical Engineering and Naval Architecture, University of Split Maximum entropy production
(MEP) principle has been derived recently in a ground‑breaking work of
Dewar1, who used an information theoretical formulation of non‑equilibrium
statistical mechanics. The MEP principle states that the MEP state is
reproducibly selected because it is the most probable non‑equilibrium
steady state compatible with given external constraints. In the linear region, for
the case of an electric network, MEP is equivalent to the Kirchhoff’s
loop law when overall energy conservation is assumed2. In the case
of heat conduction in an anisotropic crystal, MEP is equivalent to the
Onsager-Rayleigh principle of the “least dissipation of energy”3.
In the nonlinear region, MEP provides generalization of Onsager’s
theory for situations and systems far from equilibrium. For instance, when
modeling global climate MEP predicts that winds and currents driven by
thermal gradients establish themselves as the most effective heat transport
from the warmer tropics to the colder poles which maximizes the entropy
production. We shall discuss what
would be the best mode of MEP application in biochemistry4,5 and
how realistic are selected states with maximum information entropy and
maximum dissipation. Natural selection for the coexistence of ordered and
dissipative regions far from equilibrium can be understood as an expression
of the same basic concept, namely, selection of the most probable state,
which produces and exports more entropy to the environment than a purely
dissipative 'soup'. To conclude, recent research at the 1. R.C. Dewar (2005) Maximum entropy production
and the fluctuation theorem, preprint. 2. P.
Županović, D. Juretić, 3. P.
Županović, D. Juretić, 4. P.
Županović, D. Juretić (2004) Croat. Chem. Acta 77:
561-571. 5. P.
Županović, D. Juretić (2003) Comput. Biol. Chem. 27: 541-553. |
Abstract: MATH/CHEM/COMP 2005,
|
Applicability of the SQM
Force Field Method
to the Vibrational Spectra
of charged systems:
Sodium Acetate
Gábor Keresztury1, Krisztina István1 and Tom Sundius2 1Chemical Research Center, Hungarian Academy of Sciences, P.O. Box 17, H-1525 Budapest, Hungary 2Department of Physical Sciences, University of Helsinki, P.O. Box
64, FIN-00014 Helsinki, Finland The applicability of the scaled quantum mechanical
force field (SQM FF) method1,2 to the prediction of the
vibrational spectra of a charged molecule has been studied by the example of
the acetate ion (CH3CO2–) in sodium
acetate for which an efficient empirical valence force field (SVFF) based on
observed IR spectra of six isotopomers of sodium acetate is available in the
literature. Standard SQM FF calculations done on a free acetate ion at the
B3LYP/6-31G* level failed to give an acceptable estimation of even the most
characteristic features of the observed spectra, which can be exemplified by
the gross overestimation of the frequency separation of the naCO2–
and nsCO2– vibrations.
In search for a better description, SQM calculations
were done for three simple structural models of sodium acetate, testing
different QM methods. The results indicate that in addition to taking into
account the dielectric field effect of the surrounding medium, incorporation
of a Na+ counterion is necessary to achieve a realistic simulation
of the IR and Raman spectra. Satisfactory results were obtained with a
bidentate Na-acetate complex by the SQM method coupled with a continuum model
at the B3LYP/6-31+G** level, whereas the use of the Onsager-type spherical
cavity model and the polarizable continuum model (PCM) were found preferable
over SCI-PCM.
Acknowledgements. The authors are grateful to Dr. O. Berkesi ( 1. P. Pulay, G. Fogarasi, G.
Pongor, J.E. Boggs, A. Vargha (1983) J. Am. Chem. Soc. 105: 7037. 2. G. Fogarasi, X. Zhou, P.W.
Taylor, P. Pulay (1992) J. Am. Chem. Soc. 114: 8191. |
Abstract: MATH/CHEM/COMP 2005,
|
Gas phase
reactions of ambiental CO and SO2 with ozone
Nenad Kezele1,
Glenda Šorgo1, Leo
Klasinc1,2 and
William A. Pryor2 1Ruđer Bošković Institute, POB 180, HR-10002 2Biodynamics Institute, Scientific
cooperation with Biodynamics Institute exists now for more than a decade and
is documented by several joint publications1-6. Thus,
peroxynitrite and peroxynitrous acid, nitrogen oxide anions and nitrogen
oxyradicals, toxicity of ozone and synergistic effects of nitrogen oxides and
other oxidants as well as biological and health effects of air pollution were
addressed. The research part in Zagreb is more oriented to the gaseous phase
and atmospheric measurements. Here we report the influence of elevated
atmospheric ozone concentrations on readings of ambiental CO and SO2
pollution e.g. from combustion
sources. 1. L. Klasinc, D. Srzić, Lj. Paša-Tolić,
S. Martinović (1996) Gas Phase Properties of ONOO-anion and
ONOO-radical. Croat. Chem. Acta 69: 1007-1011. 2. B. Juršić, L. Klasinc, S. Pečur, W.A.
Pryor (1997) On the Mechanism of HOONO to HONO2 Conversion. Nitric Oxide Biol. - Ch. 1: 494-501. 3. M. Friedman, S. Kazazić, N. Kezele, L. Klasinc, S.P. McGlynn, S. Pečur, W. A. Pryor (2000) Role of Nitrogen Oxides in Ozone Toxicity. Croat. Chem. Acta 73:
1141-1151 4. S. Kazazić, S.P. Kazazić,
L. Klasinc, S.P. McGlynn, W.A. Pryor (2002) Proton Affinities of N-O Anions
and their Protonated Forms. J. Phys.
Org. Chem. 15: 728-731. 5. S.P.
Kazazić, L. Klasinc, S.P. McGlynn, W.A. Pryor (2004) Proton Affinities of Nitrogen Oxyradicals.
Croat. Chem. Acta, 77: 465-468. 6.
T. Cvitaš, L. Klasinc, N. Kezele, S.P. McGlynn, W.A. Pryor (2005) How
Dangerous is Ozone? Atmos. Environ.
(in press). |
Abstract: MATH/CHEM/COMP 2005,
|
|
A New Approach to NMR Chemical Shift Additivity Parameters Using Simultaneous Linear Equation Method Rabah A. Khalil and Yosif A. Shahab Department
of Chemistry, A new approach to nmr chemical shift additivity
parameters using simultaneous linear equation method has been introduced.
Three general 15N nmr
chemical shift additivity parameters with physical significance for 193
compounds of aliphatic amines in methanol and cyclohexane and their
hydrochlorides in methanol have been derived. A characteristic feature of
these additivity parameters is the individual equation can be applied to both
open‑chain and rigid systems. The factors that influence the 15N
chemical shift of these substances have been determined. A new method for
evaluating conformational equilibria at nitrogen in these compounds using the
derived additivity parameters has been developed. Conformational analyses of
these substances have been worked out. In general, the results indicate that
there are four factors affecting the 15N chemical shift of
aliphatic amines, paramagnetic term (p-character),
lone pair-proton interactions, proton-proton interactions, and molecular
association. |
Abstract: MATH/CHEM/COMP 2005,
|
Electronic
Structure of
halogenated
diphenylmethanones Leo Klasinc1,
Berta Košmrlj2,
Branka Kovač1 and Boris Šket2 1Ruđer Bošković Institute, Bijenička 54, 2University of HeI photoelectron spectra
of a series of 2-halo-substituted 1,3-diphenyl-1,3-diketones have been
measured. The assignment of the spectra was made by comparison with
photoelectron spectra of related compounds and by density functional theory
calculations with the B3LYP hybrid functional. The effect of halogen
substitution is discussed, and the results are correlated with the data
obtained from their uv spectra.
Namely, dibenzoylmethanes are known to exhibit strong absorption in the uv region and almost no fluorescence,
which makes them good photostabilization agents.

Figure 1. 2-F 1,3-diphenyl-1,3-diketone. 1. S. Tobita, J. Ohba, K. Nakagawa, H. Shizuka (1995) J.
Photochem. Photobio. A 92:
61-67. 2. S.S. Kim, J.S. Lim, J.M. 3. B.
Košmrlj, B. Šket (2000) J.
Org. Chem. 65: 6890-6896. 4. T. Pasinszki, T. Veszpremi, M. Feher,
B. Kovač, L. Klasinc, S.P. McGlynn (1992) Int. J. Quantum. Chem. Symp. 26:
443-453. |
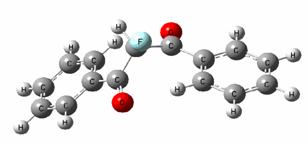
Abstract: MATH/CHEM/COMP 2005,
|
|
Metal Complexation of
Thiacrown Ether Macrocycles by Mass Spectrometry in Liquid and Gas Phase Leo Klasinc1, Bogdan Kralj2, Kata Mlinarić-Majerski1,
Marko Rožman1,
Dunja Srzić1,
Ines Vujasinović1 and Dušan Žigon2 1Ruđer Bošković Institute, HR-10002 2 Jožef Stefan Institute, SI-1000 Crown and thiacrown ethers
are an important and widely used class of organic molecules particularly for chemical
separations because of their selective binding of metal ions. Here we
investigate the complexation of Cu, Cd and Hg cations with following
structurally related thiacrown ether macrocycles:
in the liquid phase (fast
atom bombardment, FAB, and electrospray ionization1, ESI) and gas phase (laser desorption/ionization, LDI) mass spectrometry.
Special attention was paid to selectivity of formation and stability of the
monomeric products, as well as to the addition of a second crown ether
molecule. 1. S.M. Williams, J.S. Brodbelt, A.P. Marchand, D. Cal, K. Mlinarić-Majerski (2002) Anal. Chem. 74: 4423-4433. |



Abstract: MATH/CHEM/COMP 2005,
|
Data mining and identification of
drug side-effects during clinical trials
Paško Konjevoda Ruđer
Bošković Institute, POB 180, Bijenička 54, HR-10002 Clinical trials are
important part of new drug testing. However, side-effects are common finding
during clinical trials. The type and level of observed side-effects must be
precisely defined and explained. Data mining techniques are efficient and
robust systems capable of proactively gathering informations about the safety
of new drugs. In our presentation we explain the concrete example of
identification and explanation of side-effects during bioequivalence testing,
and capability of data mining techniques to analyse this data and use it more
effectively in comparison to classical statistics. |
Abstract: MATH/CHEM/COMP 2005,
|
|
Progressive Multiple Sequence Alignments from Triplets Matthias Kruspe and Peter F. Stadler Bioinformatics
Group, Department of Computer Sciences, The quality of progressive
sequence alignments strongly depends on the performance of the pairwise alignment
steps which are necessary to obtain the final alignment. The correct
identification of insertions or deletions that occurred during the
evolutionary history of the taxa considered is crucial. This becomes
fundamental particularly for rather divergent nucleotide sequences where the
information content within the sequences is low. The pairwise examination of
the sequences thus is not satisfactory in many cases. We use a natural gap cost model which deals with all different variations of gap openings or extensions for three sequences. Gap penalties are adjusted to global and local sequence properties. The phylogenetic history of the sequences is represented by a network which also determines the order of the three-way alignments. To speed up computation time and limit memory equirements a divide-and-conquer approach is used. We find the obtained alignments more reliable compared to alignments built with other alignment tools such as ClustalW. Because of the lesser information content compared to amino acid sequences, the benefit of our tool applies especially to RNA sequences. Furthermore we figure out that the natural gap costs model has a benefit compared to the quasi-natural gaps costs used in other algorithms. |
Abstract: MATH/CHEM/COMP 2005,
|
Geometric
and electronic structure of
carbon
nanotube junctions
István László Department of Theoretical Physics, The electronic properties
of carbon nanotubes are usually obtained with the help of the zone folding
method, which is based on the graphene electronic structure. The
rolling up of the graphene changes, however, the angles and distances
between the carbon atoms in the hexagonal network and in a more sophisticated
calculation curvature effects and the corresponding hybridizations must
be taken into count. The electronic structure is modified also by possible
non hexagonal polygons in the hexagonal network. This is the case if we
study the electronic properties of a nanotube junction. Nanotube junctions
are possible candidates for building blocks in nanoscale electronic devices
and in an ideal case each nanotube junction must contain at least six
heptagons if other polygons are not allowed. If the chirality of the tubes of
a junction is given there are also various possibilities for the positions of
the heptagons and each of them must have different electronic properties. In
this work the connection between the geometric and electronic structure of a
junction will be studied using various tight-binding methods. |
Abstract: MATH/CHEM/COMP 2005,
|
|
An Introduction to Nonuniform Random Variate Generation Josef
Leydold Department for Statistics and Mathematics,
University of Economics and Business Administration, Augasse 2-6, A-1090 Wien,
Austria This short course gives an
comprehensive survey on nonuniform random variate generation. Besides basic
principles for the univariate case we also describe methods for sampling from
multivariate distributions. The emphasis of the course will be on so called
automatic methods (also known as black box algorithms). |
Abstract: MATH/CHEM/COMP 2005,
|
|
ENUMERATION
OF KEKULÉ STRUCTURES IN GRAPHITE István Lukovits Surface
Modification and Nanostructures, Graphite seems to be the
most "aromatic" structure, because it is an infinitely large,
peri-condensed, benzenoid. Obviously the enumeration of the Kekulé
structures in graphite cannot be accomplished by applying constructive
methods. In this work various algorithms were proposed in order to determine
the number of Kekulé structures in different graphite sheets. Based on
the techniques used to enumerate Kekulé structures, the number of
conjugated circuits was also determined. The results indicate that the numerical
value of the resonance energy per electron (REPE) strongly depends on the
shape of the actual graphite sheet. The values of the REPE as obtained for
graphite sheets were compared with values obtained for nanotubes and
nanobelts. The latter structures seem to be somewhat less
"aromatic" than infinite graphite. 1. I. Lukovits (2004) Resonance energy in
graphite. J. Chem. Inf. Comput. Sci.
44: 1565-1570. 2. |
Abstract: MATH/CHEM/COMP 2005,
|
|
Stabilizing Residues in Proteins Csaba Magyar1, Michael M. Gromiha2 and István Simon1 1Institute of Enzymology, Karolina 29, 2CBRC AIST, 2-42 Aomi, Koto-ku, Stabilizing Residues (SRs)
are defined by combining several methods based mainly on the interactions of
a given residue with its spatial, rather than its sequential neighborhood and
by considering the evolutionary conservation of the residues. SRs are
expected to play key roles in the stabilization of proteins. A residue is
defined as a Stabilizing Residue if it is highly conserved, it has high
Surrounding Hydrophobicity, high Long Range Order values and it belongs to a |
Abstract: MATH/CHEM/COMP 2005,
|
Application of Random Matrix Theory
to BioSignal Analysis
Mladen Martinis and Vesna Mikuta-MartinisTheoretical
Physics Division, Ruđer Bošković Institute, Zagreb, Croatia
Some characteristic
properties of fluctuating biosignals are compared with the predictions of the
Random Matrix Theory (RMT). The aim is to connect the structures of these
fluctuations with the universality classes of RMT, in order to distinguish
healthy and unhealthy states of the human body. In particular, the
statistical properties of random matrices with critical spectra will be
considered, as well as the properties of random determinants and random
distances between eigenvalues. A possible application of the RMT to ECG, EEG
signals and to DNA microarray data will be indicated. |
Abstract: MATH/CHEM/COMP 2005,
|
|
Non-vibrational
features in NIR FT-Raman spectra of
lanthanide sesquioxides Zlatko Meić and Tomislav Biljan Faculty
of Science, Department of Chemistry, Strossmayerov trg 14, 10000 Zagreb,
Croatia The majority of lanthanide
sesquioxides and yttrium sesquioxide show additional bands in FT-Raman
spectra1 (after the excitation with the 1064 nm line of a Nd:YAG
laser) that cannot be explained by vibrational origin. Additional bands in
the FT-Raman spectra of heavy lanthanide sesquioxides appear in the Stokes
region of the spectrum, but there are also some very strong unexpected bands
in the anti-Stokes region of some light lanthanide sesquioxides and in
yttrium sesquioxide, notably around 800 and 1100 cm-1. The
non-vibrational bands observed in FT-Raman spectra of lanthanide sesquioxides
and yttrium sesquioxide are not seen in Raman spectra excited in the visible
region. A possible origin of these additional bands is in luminescence of
lanthanide ions. Strong anti-Stokes bands observed in FT-Raman spectra of Y2O3,
La2O3, Gd2O3 and Lu2O3
are explained by NIR luminescence of Yb3+ impurities present in sesquioxides
after the excitation with the 1064 nm line of a Nd:YAG laser.
Figure 1. FT-Raman spectra of some lanthanide sesquioxides. 1. T. Biljan, S. Rončević, Z. Meić, K.
Kovač (2004) Chem. Phys. Lett. 395: 246–252. |

Abstract: MATH/CHEM/COMP 2005,
|
CASE
via MS:
Generation of Molecular Structures and Structure Ranking by Mass Spectra
Markus Meringer Department of Medicinal Chemistry, Kiadis B.V., Zernikepark 6-8,
9747 AN Computer-aided
structure elucidation (CASE) is of immense importance for present-day drug
discovery programs. Thanks to modern screening methods a large number of
biologically active compounds can be found in a short time. Structure elucidation
then becomes a serious bottleneck in the drug discovery workflow. Due to its
high sensitivity mass spectrometry is still one of the analytical key methods
for elucidation of unknown structures. The
following computational experiment is based on low-resolution electron impact
mass spectra of small organic compounds, taken from the NIST MS library1.
The aim was to evaluate the accuracy of a structure-ranking algorithm
included in MOLGEN-MS2. The method is based on virtual
fragmentation of a candidate structure and comparison of the fragments’
isotopic patterns against the spectrum of the unknown compound. This way a
structure-spectrum compatibility matchvalue is computed, ranging from 0 (no
match) to 1 (perfect match). Of special interest was the matchvalue’s
ability to distinguish between the correct and false constitutional isomers. Therefore
a quality score was computed in the following way: For a (randomly selected)
spectrum-structure pair from the MS library all constitutional isomers were generated
using the structure generator MOLGEN3. For each isomer the
matchvalue with respect to the library spectrum is calculated4,
and isomers are ranked according to their matchvalues. The quality of the
ranking can be quantified in terms of the relative ranking position:
This
procedure was repeated for 100 randomly selected spectrum-structure pairs
that fulfilled certain conditions (molecular mass ≤ 200 amu, 1 <
number of isomers ≤ 10000). In this first approach an average RRP of
0.30 was computed. More sophisticated algorithms for virtual fragmentation5,6
raise hope for better ranking results. In combination with more accurate high‑resolution
MS/MS techniques this could pave the way towards automated structure
elucidation via mass spectrometry. 9.
NIST/EPA/NIH Mass Spectral Library, NIST '98 version. 10. A. Kerber, R. Laue, M. Meringer, K.
Varmuza (2001) MOLGEN-MS: Evaluation of Low Resolution Electron Impact Mass
Spectra with MS Classification and Exhaustive Structure Generation.
Advances in Mass Spectrometry, Vol. 15, Wiley, pp. 939-940. 11. T. Grüner, A.
Kerber, R. Laue, M. Meringer (1998) MOLGEN 4.0. MATCH
Commun. Math. Comput. Chem. 37: 205‑208. 12. M. Meringer (2004) Mathematical
Models for Combinatorial Chemistry and Molecular Structure Elucidation. Logos‑Verlag
Berlin (in German). 13. MassFrontier 4.0. HighChem, Ltd., 14. ACD/MS Fragmenter. Advanced Chemistry Development, Inc., |
Abstract: MATH/CHEM/COMP 2005,
|
ESTIMATION OF STABILITY OF DIAMINOETHANE COMPLEXES BY THE OVERLAPPING SPHERES METHOD WITH
A VARIABLE CENTRAL SPHERE RADIUS
Ante MILIČEVIĆ and Nenad RAOS Institute
for Medical Research and Occupational Health, HR-10001 The method of
overlapping spheres (OS) for the estimation of the stability constants of
coordination compounds is based on the calculation of the overlapping volume,
V*, of the central sphere (situated at the
central or a bite atom) and the van der Waals spheres of surrounding atoms.
The most critical parameter in the model is the radius of the central sphere,
Rv, which is usually taken to be 3 or 4
Å. Here we
propose the model with variable value of the Rv parameter,
making it to correspond to its value at the maximum of the volume density,
V*(Rv)/ (4/3 Rv3 π), for each molecule. For the
training set (N = 14) the range of maximum volume densities and Rv are 0.556
– 0.722 and 2.7 – 3.0 Å, respectively. Linear
regressions of stability constants (log K1) for mono-complexes of diamines with copper(II) and nickel(II) measured at 0
and 25 oC on overlapping spheres volume, V*, yielded slightly, but significantly better results than
calculations done at Rv = 3 Å. The four regression yielded <r> = 0.786 (r = 0.677 – 0.858) and <r> = 0.796 (r = 0.681 – 0.878) for the constant and variable Rv value,
respectively. Also, test on five copper(II) mono-complexes (25 oC), not included in the training set, gave
residuals in the range 0.01 – 0.71 (r.m.s. = 0.33) log K
units, which is slightly
better than the results for Rv = 3 Å (range = 0.02 – 0.75, r.m.s. = 0.34 log K units), but substantially better than results obtained with Rv = 4 Å (range
0.15 – 0.91, r.m.s. = 0.48 log K units). In spite of the slight differences, which should be mostly
attributed to Rvvar ≈ 3Å, the proposed approach seems to be sound for the
further improvement of the model. |
Abstract: MATH/CHEM/COMP 2005,
|
Computational
identification of ncRNAs in Invertebrates by comparative genomics
Kristin Missal1,
Dominic Rose1 and
Peter F. Stadler1,2 1Bioinformatics Group, Department of Computer Science, 2Institute for Theoretical
Chemistry, The analysis of animal
genomes showed that only a minute part of their DNA codes for proteins. Recent experimental results agree,
however, that a large fraction of these genomes is transcribed and hence is
probable functional at the RNA level1. A computational survey of
vertebrate genomes had predicted thousands of previously unknown ncRNAs with
evolutionary conserved secondary structures4. To extend this
comparative studies to invertebrates is difficult, since most ncRNAs evolve
relatively fast at the sequence level while conserving their characteristic
secondary structures. Hence, independent screens in invertebrates are
necessary. Urochordates, as the sister group of vertebrates, that do not
share the genome duplications, are of particular interest in this context.
The genomes of two ascidians, Ciona intestinalis and Ciona savignyi have been
sequenced, and a third project for the larvacean Oikopleura dioica is on the
way. This gives us sufficient data and annotation to screen for evolutionary
conserved ncRNAs in urochordates independent of vertebrates. Further the
genomes of two nematodes, Caenorhabditis elegans and Caenorhabditis briggsae,
are also available providing us with an additional separate set of
prostostomia which gives us an extended spot of ncRNA evolution. Here, we report
computational screens for evolutionary conserved ncRNAs in two independent
sets of invertebrates: C. intestinalis, C. savignyi, O. dioica and C. elegans,
C. briggsae. First, we identify conserved ncDNA regions (excluding regions
annotated to be repetitive) by BLAST alignments. Conserved ncDNA regions with
short distance between are combined considering consistence checks. Global
alignments of those regions using CLUSTALW are computed. These alignments are
screened with RNAz3 to detect regions that are also conserved at
the secondary structure level. 1. J.S. Mattick (2004) RNA regulation: a new genetics?. Nat. Rev. Genet. 5(4): 316-323. 2. T.M. Lowe, S.R. Eddy (1997) tRNAscan-SE: A program for
improved detection of transfer RNA genes in genomic sequence. Nucl. Acids Res. 25: 955-964. 3. S. Washietl, I.L. Hofacker, P.F. Stadler (2005a) Fast
and reliable prediction of noncoding RNAs. Proc. Natl. Acad. Sci. USA 102:
2454-2459. 4.
S. Washietl, I.L. Hofacker, P.F. Stadler (2005b) Thousands of noncoding RNAs
with conserved structure in mammalian genomes. (in review). |
Abstract: MATH/CHEM/COMP 2005,
|
|
Symmetry Properties of Tetraammine Platinum(II)
with C2v and C4v point groups Ghorban Ali Moghani and Ali Reza AshrafiDepartment
of Mathematics, Faculty of Science, Payame Noor, University, Let G be a weighted graph with
the adjacency matrix A = [aij]. An Euclidean graph associated to a
molecule is defined by a weighted graph with the adjacency matrix D = [dij],
where for i ≠ j, dij is the Euclidean
distance between the nuclei i and j. In this matrix dii can
be taken as zero if all the nuclei are equivalent. Otherwise, one may
introduce different weights for different nuclei. Balasubramanian computed
the Euclidean graphs and its automorphism groups for benzene, eclipsed and
staggered forms of ethane and eclipsed and staggered forms of ferrocene (see Chem. Phys. Letters (1995) 232: 415.). In this work a simple
method is described, by means of which it is possible to calculate the
automorphism group of weighted graphs. We apply this method to compute the
symmetry of tetraammine platinum(II) with C2v and C4v
point groups. |
Abstract: MATH/CHEM/COMP 2005,
|
Optimizing Barbeques and Tanimoto
Scores with Applications in Regulatory Motif Discovery
Axel Mosig1,2, Sonja J. Prohaska1,2 and Peter F. Stadler1,2,3,4 1Interdisciplinary 2Bioinformatics, Department of
Computer Science, 3Institute for Theoretical
Chemistry, 4The Santa Fe Institute, Understanding
the mechanisms of gene expression is a major challenge of current genomics.
On the level of transcription, gene expression in Eukaryotes is known to be regulated
by several transcription factors binding to clusters of respective binding
sites. These binding site clusters are also referred to as cis-regulatory
modules. We propose
multiple-alignment like pattern discovery methods that allow to discover putative
regulatory modules. Our algorithms are based on detecting clustered
occurrences of binding sites that occur simultaneously in several genomic
sequences. Computationally, this is achieved through solving the so-called
best barbeque problem1 in combination with Tanimoto scores, which were
originally introduced as a measure for feature-based molecular similarity2
and have recently been proposed as a similarity measure between regulatory
modules3. Acknowledgements. This work was supported by
the DFG Bioinformatics Initiative. 1. A. Mosig, T. Biyikoglu, S. J. Prohaska, P. F. Stadler (2004) Discovering cis-regulatory modules by Optimizing Barbeques. (submitted)
2. P. Perco, A. G.
Mayer, A. Lukas, R. Oberbauer, B. Mayer, (2005) A genetic algorithm to derive joint promoter modules in coexpressed
genes. (submitted) 3. J.B.O. Mitchell (2001) The relationship between the sequence
identities of α-helical proteins in the PDB and the molecular
similarities of their ligands. J.
Chem. Inf. Comp. Sci. 41:
1617-1622. |
Abstract: MATH/CHEM/COMP 2005,
|
|
Relinking Marriages in Genealogy of Ragusan Noble Families between 12th and 16th Century Andrej Mrvar and Vladimir Batagelj Genealogies can be represented as graphs in
different ways: as Advantages and disadvantages of all three
presentations will be discussed. The emphasis will be given to all possible
relinking marriages (blood and non-blood marriages) in p‑graphs containing 2 up to 6 vertices. As an example we will take the genealogy of Ragusan
( We will compare the frequency distribution of different types of relinkings to some
other genealogies (genealogy of island Silba, genealogy of European royal
families). Analyses were done using program for analysis and
visualization of large networks Pajek, which can be downloaded for free: http://vlado.fmf.uni‑lj.si/pub/networks/pajek/default.htm |
Abstract: MATH/CHEM/COMP 2005,
|
Toxicity
of Aliphatic Ethers Revisited
Sonja Nikolić,1 Ante Miličević2
and Nenad Trinajstić1 1Ruđer Bošković Institute, P.O.B. 180,
HR-10002 2Institute of Medical Research and Occupational
Health, P.O.B. 291, HR-10001 There are in the
literature several reports on QSAR study of toxicity of aliphatic
ethers. Authors of these reports
used a variety of molecular descriptors: vertex-connectivity index c, the first-order and the second-order
valence vertex-connectivity indices 1cv and 2cv,
weighted identification number WID,
Balaban indices J and Jhet and edge-connectivity
index e. We collected all these indices
for 21 aliphatic ether and applied our CROMRsel modeling procedure.1,2 This is a multivariate procedure that
has been designed to select the best possible model among the set of models
obtained for a given number of descriptors, the criterion being the standard
error of estimate. The quality of
models is expressed by fitted (descriptive) statistical parameters: the
correlation coefficient (Rfit),
the standard error of estimate (Sfit)
and the Fisher's test (F). The models are also
cross(internally)-validated by a leave-one-out procedure. Statistical
parameters for the cross-validated models are symbolized by Rcv and Scv, where
subscript cv denotes the
cross-validation. The
best single-descriptors model is based on WID
(Rfit=0.942, Rcv=0.920, Sfit=0.14, Scv=0.16), the best
two-descriptor model is based on c and J (Rfit=0.966,
Rcv=0.951, Sfit=0.10, Scv=0.13), the best
three-descriptor model is based on 1cv, Jhet
and e (Rfit=0.981,
Rcv=0.968, Sfit=0.08, Scv=0.10) and the best
four-descriptor model is based on c,1cv, J and Jhet (Rfit=0.983,
Rcv=0.969, Sfit=0.08, Scv=0.10). These last two models are comparable
among themselves – our choice is the model with less descriptors. The best models from the literature3,4
are based on the variable connectivity index 1 cf (Rfit=0.975, Sfit=0.10), and
on Xu index5 and atom type descriptors (Rfit=0.987, Rcv=0.965,
Sfit=0.07, Scv=0.12). 1. B.
Lučić,
2. A. Miličević, 3. M. Randić, S. C. Basak (2001) J. Chem. Inf. Comput. Sci. 41: 614-618. 4. L. Xu (1996) Chemometrical
Method, Scientific Press of 5. B. Ren (unpublished). |
Abstract: MATH/CHEM/COMP 2005,
|
Tandem
repeats in three dimensional
structures
of proteins
Borut T. Oblak and Branko Borštnik National
With growing amount of knowledge about
the primary structure and three dimensional structure of proteins it is
becoming more and more evident that an appreciable fraction of proteins does
not possess well defined three dimensional structure. Such proteins usually
contain tandem repeats which are usually associated with other sequential and
functional features which are the consequence of rapid evolution on molecular
scale. We searched the amino acid sequence file of the proteins whose
structures are deposited in the Protein Data Bank for the tandem repeat
patterns. Several filamentous proteins and other proteins were found which do
not possess necessarily well defined three dimensional structure for the
entire amino acid sequence. Tandem repeats are located by rule in the regions
where increased mutability is exhibited. The repeats are the results of the
repeat amplification mechanisms which are the fundamental driving force of
molecular evolution. Slippage mutations, transpositions and related events
generate repetitious sequences in the coding regions of genes. |
Abstract: MATH/CHEM/COMP 2005,
|
A Calculation and a Representation of Edge‑Transitive
Maps of Small Order
Alen Orbanić and Tomaž PisanskiIMFM,
Jadranska 19, SI- 000 We present a calculation
of non-degenerate edge-transitive maps2 up to 100 edges which is
at the present time a computationally hard problem. A computation was
performed by use of program LOWX made by Conder and Dobcsanyi1. We
have built in an optimization into the procedure for searching factor groups up to some order, which
allows exclusion of factor groups having some forbidden relations and thus
optimizing a running time of the algorithm for up to 50%. Since these maps have a
high symmetry, we consider several automated procedures to draw their
graphical representations with highly symmetric drawings3. Good
drawings are a very useful tool in the research of the maps. Some chemical structures,
like fullerenes, can be described as maps of high symmetry. A calculation and
determination of such maps can yield to better understanding of topology of
some chemical structures. 1. M. Conder, P.
Dobcsanyi (2001) Determination of all regular maps of small genus. J. Comb. Theory B 81: 224-242. 2. J.E. Graver, M.E.
Watkins (1997) Locally finite, planar, edge-transitive graphs. Mem. Am. Math. Soc. 126: 601. 3. T. Pisanski et
al. Project Vega, http://vega.ijp.si |
Abstract: MATH/CHEM/COMP 2005,
|
Constructing
convex polyhedra
Igor Pak Department
of Mathematics, MIT, We investigate the problem
of constructing polyhedra from their edge length. In a joint work with Fedorchuk we obtained
sharp upper bounds for the degrees of Sabitov polynomials having the diagonal
lengths as its roots. Knowing all the diagonal lengths one can easily
construct the polyhedron. We then ask about flexible polyhedra and their
structure, discuss several constructions and how they can be
generalized. Acknowledgements. We would like to thank Bob Connelly and
Maksym Fedorchuk for helpful discussions. The author received funding from
the NSF. V. Alexandrov (1995) New example of a
flexible polyhedron, Sib. Math. J. 36: 1215-1224. B. Connelly (1979) The rigidity of
polyhedral surfaces, Math. Mag. 52: 275-283. M. Fedorchuk, I. Pak (2005) Rigidity and
polynomial invariants of convex polytopes Duke
Math. J. (to appear). I. Pak (2005) The area of cyclic polygons:
recent progress on Robbins conjectures Adv.
Applied Math.(to appear). I. Kh. Sabitov (1998) The volume as
metric invariant of a polyhedra Disc.
Comp. Geom. 20: 405-425. |
Abstract: MATH/CHEM/COMP 2005,
|
An effective potential for the natural
spinorbitals
Katarzyna Pernal1,2 1Section Theoretical Chemistry, Vrije Universiteit, De Boelelaan
1083, 1081 HV 2Institute of Physics, It is well known that in
the Kohn-Sham (KS) formulation of the density functional theory the
variational principle leads to one-electron equations for the KS orbitals with
the effective local potential. Until now it has not been clear if an
analogous effective potential exists for the natural spinorbitals and how to
construct it explicitly. This problem is of great importance for the density
matrix functional theory (DMFT), where the total energy is expressed in terms
of the one-electron reduced density matrix (1-matrix). It will be presented how
to construct an effective nonlocal potential that, for a given set of
occupation numbers, produces an optimal set of natural spinorbitals. A
problem of its nonuniqueness in the case of degenerate 1-matrices will be
discussed. If the Hamiltonian is perturbed with a static perturbation, one‑electron
equations for the natural spinorbitals together with the Lagrange equations
for the natural occupancies serve as a basis to derive coupled-perturbed
equations that yield a response of the 1-matrix to a perturbation. This leads
in a straightforward manner to calculations of the second-order response
properties of a system described by a given density matrix functional. Some
results of the dipole-dipole polarizabilities will be shown for the recently
proposed density matrix functionals. |
Abstract: MATH/CHEM/COMP 2005,
|
|
Einstein: from Special Theory of Relativity (1905),
through General Theory of Relativity (1916), until Cosmological Term (1917) Krunoslav Pisk Ruđer Bošković Institute, POB
180, HR-10002 Zagreb, Croatia 1905 – Special Theory of Relativity (STR) About uniform motion in
empty space and relative motions with uniform translations among frames of
reference. The speed of light is constant in inertial frames. A new concept
of space and time: space-time continuum as an integral entity. Lorentz
transformations as coordinate links among different frames. Relations among
observables. Relativity of lengths and time intervals. Equations which
express physical laws are form-invariant (covariant) against Lorentz
transformations. A new relation between mass and energy, E = mc2, emerges naturally. 1916 – General Theory
of Relativity (GTR) Extension of STR by
imposing that equations of law of physics are form-invariant against general
space-time transformations (general covariance), which also include
coordinate relations among accelerated frames. Accelerated frames and
gravitational fields (principle of equivalence). Gravitation and curvature of
space. Equations of GTR: a)
Einstein’s field equations - energy (mass) – distribution and
corresponding pressures, as a source of curved space; b)
Einstein’s equations of motion – motion of material particle in
gravitational field. GTR and 1917- Cosmological Term When applied to the
Universe, solution of field equations of GTR are not static. Einstein
modifies original equations by introducing a cosmological term (cosmological
constant today) - λ,
which is consistent with relativity
postulates. Now, a quasi-static distribution of matter in Universe is
possible. The Universe appears a 3-dimensional curved space (with constant
radius of curvature Epilogue Thirteen years ago (1930),
after Hubble’s discovery of Universe expansion Einstein said that
introduction of cosmological term was “the biggest blunder of my
life”. Recent measurements
indicate that Universe accelerates and abandoned cosmological term could play
important role – “dark energy”4. 1. A. Einstein (1905) On the Electrodynamics of
Moving Bodies, Annalen der Physik 17: 891. 2. A. Einstein (1916) The Foundation of the GTR, Annalen der Physik 49: 769. 3. A.Einstein (1917) Cosmological Considerations
of GTR, Sitzungsberichte der
Preussischen Akad. d. Wissenschaften. 4. P.J.E. Peebles, B. Ratra (2003) Rev. Mod. Phys. 75: 607. |
Abstract: MATH/CHEM/COMP 2005,
|
Variable
topological indices in drug design
Matevž
Pompe1 and 1University of Ljubljana, Faculty of Chemistry and Chemical Technology,
Aškerčeva 5, 10000 Ljubljana, Slovenia 2National Institute of Chemistry, Hajdrihova 19, 1000 Ljubljana, Slovenia Quantitative
structure-activity relationship (QSAR) studies have been successfully applied
for the modeling of different important biopharmaceutical properties. During
the first stage of drug design large data bases are usually screened for
suitable candidates. In order to complete screening in the reasonable time a
simple structural representation should be used which at the same time
enables creation of reasonably accurate prediction models. Variable
topological indices can be considered as good candidates because they are
easily calculated, give good prediction models and enables structural
interpretation of the obtained models. Several modifications of variable
topological indices will be presented to account for modeling of large
molecules as well as compounds where certain part of the molecule suppresses
the modeled activity. |
Abstract: MATH/CHEM/COMP 2005,
|
|
MOLECULAR
BASIS OF LFER. SIMPLE MODEL FOR THE
ESTIMATION OF BRÖNSTED EXPONENT IN ACIDOBASIC CATALYSIS Robert Ponec A simple model was
proposed1 allowing to estimate the Brönsted exponents in acidobasic
catalysis on the basis of the pK
values of the species participating in the proton transfer process. The
approach was tested using the experimental data on the basically catalysed
halogenation of carbonyl compounds and on the proton removal from
nitroalkanes. It has been shown that the model is able to reproduce the
Brönsted exponents not only in the case of "ordinary"
Brönsted plots with the slope within the expected range 0 – 1 but
also for unusual plots with negative slope. In addition to this the proposed
model opens the possibility of the calculation of the activation energies of
a given proton transfer reaction and also provides straightforward
theoretical justification for the validity of R. Ponec (2004) Collect.
Czech. Chem. Commun. 69: 2121. |
Abstract: MATH/CHEM/COMP 2005,
|
|
Rearrangement of Regulatory Elements Sonja
J. Prohaska1,2, Axel Mosig1,2 and Peter F. Stadler1,2,3.4 1Interdisciplinary
Center of Bioinformatics, University of Leipzig, Germany 2Bioinformatics,
Department of Computer Science, University of Leipzig, Germany 3Institute
for Theoretical Chemistry, University of Vienna, Austria 4The Santa Fe Institute,
Santa Fe, New Mexico, USA "Phylogenetic footprinting"1 is the latest method to
find regulatory elements, which are conserved in non-coding sequences around
orthologous and functionally equivalent genes. Therefore, these elements can
be detected by a comparative genomic approach based on sequence alignment.
The result is a set of conserved 'footprints' that are in the same order and
orientation in at least two of the input sequences. Since regulatory elements
are rather insensitive to shuffling of single modules, putative protein
binding sites might be missed by a simple comparative approach. We expanded
the footprinting approach and look for rearranged footprint clusters,
disregarding the order and orientation of the individual motifs. The main
purpose of this contribution will be to interpret the biological relevance of
this approach when applied to the duplicated Hox cluster sequences, that show
a high rate of footprint remodeling. We detect rearranged footprint clusters by solving
the best barbeque problem2 for a given set of motifs and a set of
orthologous sequences, that are supposed to share a common set of motifs.
This means, we use bbq to look for the maximal set of footprints within a
cluster of defined length L that occurs in all N sequences. The
required set of motifs is either taken from transfac matches to the sequences
or decomposed footprint clusters. An application to the duplicated Hox cluster
sequences – up to eight in teleost fishes and four in all other
vertebrates – will shed light on the importance of methods like bbq for
the detection of reorganized regulatory elements and rise ideas on the
dynamics of structural organization of these elements. Acknowledgements. This work was supported in part by the DFG
Bioinformatics Initiative. 1. S.J. Prohaska, C. Fried, C. Flamm, G.P.
Wagner, P.F. Stadler (2004) Mol. Evol. Phylog. 31: 581-604
. 2. A. Mosig
(2005) A tool for discovering regulatory modules using weighted barbeques.
(submitted). |
Abstract: MATH/CHEM/COMP 2005,
|
|
Graphical Representation of Proteins as Four-Color Maps
and Their Numerical Representation
|
Abstract: MATH/CHEM/COMP 2005,
|
THE
MEANING OF CLUSTER AND ITS INFORMATION CONTENT
Guillermo RESTREPO Laboratorio de
Química Teórica, Universidad de Pamplona, Pamplona, Colombia A general problem of the studies of Cluster
Analysis (CA) is the one related to the selection of the clusters in the
dendrogram (tree). There are several methods for selecting clusters in a
tree, some of them are the “Phenon line”1, the
amalgamation coefficient2 and some others. However, the most common method in
order to look for clusters is the Phenon line but it is a subjective
procedure since it includes the previous knowledge of the researcher
regarding the set Q under study. On
the other hand, taking into consideration the uses of CA as a tool for
determining similarity relationships among the elements of Q, we consider that a criterion for
selecting the clusters of a dendrogram should include the similarities among
the elements3. In order to build up a method for selecting
clusters in a tree we developed two mathematical procedures free of
subjectivities. Both of them are based on the chemotopological concept of
maximal n-subtree4. This
concept allows selecting a particular sort of branches (clusters) of the
dendrogram for a given value of Acknowledgements.
We thank Dr. Mesa from
the Universidad del Valle ( 1. P.H.A.
Sneath, R.R. Sokal (1973) Numerical Taxonomy: The Principles and Practice
of Numerical Classification., W.H. Freeman Comp., San Francisco, p. 573. 2. Q. Liao, B. Manteuffel, C. Paulic, D.J. Sondheimer
(2001) Emot. Behav. Disor.
Spring. 3. G.
Restrepo, E.J. Llanos, J.L. Villaveces (2005) In: Fourth Indo-US Workshop on Mathematical Chemistry (S. Basak, D.K.
Sinha, eds.), University of Pune, Pune, India, pp. 39-40. 4.
G. Restrepo, H. Mesa, E.J. Llanos, J.L. Villaveces (2004) J. Chem. Inf. Comput. Sci. 44: 68-75. 5.
S. Lipschutz (1965) General Topology,
6. C.E. Shannon (1948) |
Abstract: MATH/CHEM/COMP 2005,
|
|
The Preparation of Some Novel Indazole Derivatives by Using Chalcones Javad Safaei-Ghomi and Zohreh Alishahi Department
of Chemistry, Faculty of Science, Indazole
and its derivatives have little biological significance and have not been
found in natural products due to the difficulty for living organisms to
construct an N-N bond. Indazole derivatives exhibit variety of
pharmacological properties such as anti-inflammatory, antidepressant,
antitumor, antiarthritic and analgesic activities1. Different
synthetic pathways generate these compounds. For instance, ring closure of
pyrazole moiety, addition of hydrazine derivatives to carbonyl compounds2
and cycloaddition reaction3. Herein, we report the synthesis of
some new indazole derivatives by using chalcones as starting materials. In this procedure chalcones were reacted with
ethyl acetoacetate in a convenient method for the Michael addition followed
by intramolecular aldolization catalyzed by K2CO3 under
ultrasound irradiation. This procedure brings advantages such as mild
reaction conditions, shorter times and reducing of starting materials.
Reaction of prepared 3,5-diaryl-6-ethoxycarbonyl-2-cyclohex-1-one derivatives
1 with hydrazine hydrate or phenyl hydrazine gave corresponding indazole
derivatives 2. The structures of the synthesized compounds were assigned on
the basis of spectral data.
1. P.G. Balardi et al., Synthesis
1997: 1140. 2. E.B. Usova, L.I. Lysenko, G.D. Krapivin (2000) Molecule 5: M 128. 3. S. Matsugo, A. Takamisawa, Synthesis
1983: 852. |

Abstract: MATH/CHEM/COMP 2005,
|
De Revolution VII Six Dimensions
James Sawyer
Six Dimension Design
|
Abstract: MATH/CHEM/COMP 2005,
|
|
Software implementation for covariance NMR spectroscopic investigations Wolfgang Schöfberger1, Vilko Smrečki2
and Norbert Müller1 1Johannes Kepler University Linz, Institute of Organic Chemistry,
Altenbergerstraße 69 4040 Linz,
Austria 2Ruđer Bošković Institute, NMR-Center, Bijenička c.
54, HR-10002 Zagreb, Croatia Covariance nuclear
magnetic resonance (NMR) spectroscopy1,2 provides an effective way
for establishing nuclear spin connectivity in molecular systems.
The method, which identifies correlated spin networks in terms of
covariance between 1D traces, facilitates delineation of spin
networks and allows a probable interpretation of correlation reliability. We
have implemented software that integrates into the routine workplan. The
application of covariance NMR spectroscopy applied to HMBC experiments of
novel carbohydrate derivatives is presented. Acknowledgements. This work was supported by the Austrian Science
Fund, project P15380, the Austrian-Croatian Joint Research Project
91102/03-06 (ÖAD-Project 13/04), and the Ministry of Science, Education
and Sport of the Republic of Croatia Project 0098059. 1. R. Brüschweiler, F. Zhang (2004) Covariance
nuclear magnetic resonance spectroscopy, J. Chem. Phys. 120 (11): 5253-5260. 2. R. Brüschweiler
(2004) Theory of covariance nuclear magnetic resonance spectroscopy, J. Chem. Phys. 121 (1): 409‑414. |
Abstract: MATH/CHEM/COMP 2005,
|
|
Algebraic Kekulé Structures and Bond Orders in Benzenoids Jelena Sedlar1, Ivana Anđelić2, Damir Vukičević3, Ante Graovac2, 3 and Ivan Gutman4 1Faculty of Civil Engineering,
Matice Hrvatske 15, HR-21000 2Ruđer Bošković Institute,POB
180, Bijenička c. 54,
HR-10000 3Faculty of Natural Sciences, Mathematics and
Education, Nikole Tesle 12, HR-21000 4Faculty of Science, POB 60, 34000 Recently, a new
interpretation of the Kekulé structures (KS) in benzenoid systems was proposed.
A new quantity, Algebraic Kekulé Structure (AKS), that counts how many
p electrons for a given KS belong to each hexagon has
been defined1. In this way a possibility to characterize hexagons
numerically was offered for the first time. By
knowing the AKS for all Kekulé structures gives us a chance to define
a new, AKS bond order2. This new order is compared with the
classical Pauling bond order and experimentally measured bond lengths in a
series of benzenoid systems. Preliminary results show that both bond orders
are approximately of the same quality. 1. I.Gutman, D.
Vukičević, A. Graovac, M. Randić (2004) J. Chem. Inf. Comp. Sci. 44:
296-299. 2. J. Sedlar, I.
Anđelić, D. Vukičević, A. Graovac, I. Gutman (in
preparation). |
Abstract: MATH/CHEM/COMP 2005,
|
|
Intrinsically Unstructured Proteins István Simon H-1518 There is fast growing evidence that the unstructured
state, common to all living organisms, is essential for basic cellular
functions; thus it deserves to be recognized as a separate functional and
structural category within the protein kingdom. In this lecture recent findings
in this area are surveyed including some our recent work about preformed
structural elements involved in partner recognition by intrinsically
unstructured proteins, the way pairwise energy content estimated from amino
acid composition discriminates between folded and intrinsically unstructured
proteins and about our web server, IUPred, for the prediction of
intrinsically unstructured regions of proteins based on estimated energy
content. |
Abstract: MATH/CHEM/COMP 2005,
|
RNA Consensus Structures in
Molecular Morphology
Roman R. Stocsits1
and Peter F. Stadler1,2 1Interdisciplinary Centre for Bioinformatics, Härtelstrasse
16-18, D-04107 2Bioinformatics Group, Department of Informatics, Functional
non-coding RNA molecules perform vital functions in the cell and are a major
source of data in molecular phylogenetics. But the peculiarities of RNA
evolution, and in particular the long term conservation of their secondary
structures, have been taken into account only recently. Because the functions
of these molecules are mediated by their structures, selective pressure acts
on these structures. But structural conservation is not necessarily mediated
by sequence conservation: Many different sequences can fold into the same
(functional) structure. On the other hand, only little changes in the nucleic
acid sequences may cause large changes in secondary structures. Assuming
independent evolution of each sequence position is not a good approximation
because the highly conserved secondary structure of these molecules
introduces strong correlations between the two strands of a helix. The problem
arises that variable sequences may obscure phylogenetic signals and lead to
overestimated reliability of sequence based trees and artefacts in phylogeny
reconstruction. Therefore, phylogeny reconstruction methods can be extended
to RNA secondary structure, incorporating the slower evolution of the
structural features, provided a good model for the secondary structure
elements of the RNAs. For obtaining
the best estimate of the secondary structure of each individual sequence we
use a consensus structure of the complete set of related sequences as a constraint
for the RNA folding algorithm: basic structural features that are represented
in all sequences guide the individual sequence folding process to a
stabilized, more realistic prediction.
Figure
1. Individual sequences
are folded with support by a consensus constraint, a set of basic structural
features that are present in all sequences (mitochondrial trp-tRNA in this
example). Left: wrong
prediction without constraint. Right:
correct prediction after constraint directed folding. In this talk I
present first results of our pilot-studies with the aim to utilize the
evolution of secondary structures when reconstructing phylogenies. Our goal
is a systematic investigation: Starting from mitochondrial tRNA and rRNA with
high phylogenetic information content, we plan to extend our methods to other
types of non-coding RNAs that need to be re-evaluated by explicitly taking
into account conserved functional secondary structure elements. |


Abstract: MATH/CHEM/COMP 2005,
|
Intrinsic proofs for area and
circumradius of cyclic hexagons.
Solving equations for arbitrary cyclic polygons
Dragitin SvrtanDepartment of
Mathematics, Finding formulas for the
area or circumradius of polygons
inscribed in a circle in terms of side lengths is a classical subject. For
the area of a triangle we have famous Heron formula and for cyclic
quadrilaterals we have Brahmagupta’s formula. A decade ago D. P.Robbins3
found a minimal equations
satisfied by the area of cyclic pentagons and hexagons by a method of
undetermined coefficients and he wrote the result in a nice compact form. The
method he used could hardly be used for heptagons due to computational
complexity of the approach. In another approach with two collaborators2
a concise heptagon/octagon area formula was obtained recently (not long after
D.P.Robbins premature death). This approach uses covariants of binary quintics.
It is not clear if this approach could be effectively used for cyclic
polygons with nine or more sides. A nice survey on this and other Robbins conjectures is written by
I. Pak4. In this talk we shall
present an intrinsic proof
of the Robbins formula for the
area (circumradius and area times circumradius) of cyclic hexagon based on an
intricate direct elimination of diagonals (the case of pentagon was treated in Ref. 5) and
using a new algorithm from Ref. 6. In the early stage we used computations
with MAPLE (which sometimes lasted several days!). Next we shall explain a
simple quadratic system, which seems to be new, for the circumradius and area
of arbitrary cyclic polygons based on a Wiener‑Hopf factorization of a
new Laurent polynomial invariant of cyclic polygons. Explicit formulas for
the circumradius (and less explicit for the area) of cyclic heptagons and
cyclic octagons are obtained. We hope to apply recent new resultant formulas
of Eisenbud et
al. in our approach to cyclic polygons. Acknowledgements. We would like to thank Darko Veljan and Vladimir
Volenec for helpful discussions. 1. A.F. Möbius (1828)
Über die Gleichungen, mittelst welcher aus der Seiten eines in einen
Kreis zu beschriebenden Vielecks
der Halbmesser des Kreises un die Flahe des Vielecks gefunden werden, Crelle's J. 3: 5-34. 2. F. Miller Maley, D.P. Robbins, J. Roskies, On the areas of cyclic and
semicyclic polygons, math.
MG/0407300v1. 3. D.P. Robbins (1994) Areas of polygons
inscribed in a circle, Discrete Comput. Geom. 12: 223-236, 1994. 4. I. Pak, The area of cyclic polygons: Recent
progress on Robbins conjectures, Adv.
Applied Math. (to appear). 5. D. Svrtan, D.Veljan and V. Volenec, Geometry of
pentagons: From Gauss to Robbins, math. MG/0403503. 6. D.Svrtan (2005) A new approach to rationalization
of surds. (submitted). 7. D.Svrtan (2005) Intrinsic proof of Robbins formula
for the area of cyclic hexagons.
(submitted). 8. D.Svrtan (2005) Equations for the circumradius and
area of cyclic polygons via
Wiener-Hopf factorization. Computational aspects and some new formulas. (in
preparation). |
Abstract: MATH/CHEM/COMP 2005,
|
|
Molecular recognition of the complementary peptide structures Nikola Štambuk1, Paško Konjevoda1, Nikola Gotovac2 and Biserka Pokrić1 1 Ruđer
Bošković Institute, Bijenička cesta 54, POB 180, HR-10002 2Department of Radiology, General Hospital
Požega, Osječka bb, HR-34000 Ligand-receptor peptide interaction is
investigated considering Molecular Recognition Theory and the secondary
protein structure. The theoretical concept of molecular recognition based on
the side chain polarity modelling is discussed in the context of the protein
structure and possible function. |
Abstract: MATH/CHEM/COMP 2005,
|
|
ON THE REGULARITY OF
STRUCTURAL DEGENERACY: COMPUTER-AIDED DESIGN OF SEMIREGULARLY AND IRREGULARLY
DEGENERATE REARRANGEMENTS Serge S. Tratch1, Marina S. Molchanova2 and Nikolai S. Zefirov1 1 Department of Chemistry, Moscow State
University, Moscow 119992, Russia 2 Zelinskii Institute of Organic Chemistry,
Russian Acad. Sci., Moscow 119991, Russia Degenerate transformations, i.e., chemical interconversions with
identical initial and final structures, are classified on the basis of some
permutation groups associated with the corresponding bond redistributions.
These groups are the automorphism groups of certain labeled graphs that can
be constructed from the more simple "topology
identifying"
graphs; edges of the latter graphs represent all bonds changing their
multiplicity. If the chemical nature of reaction centers is not explicitly
taken into account, then three classes of degeneracy are distinguished. For regularly
degenerate (R)
transformations, at least one permutation converting the initial structure(s)
into the final one(s) belongs to the group of the labeled graph (and
consequently to the group of the topology graph); actually, all known
degenerate rearrangements belong just to this class. In the case of semiregular
(S ) or irregular
(I
) degeneracy, no "producing"
permutations belong to group of the labeled or both graphs, respectively.
Explicit consideration of the chemical nature of participating atoms is shown
to result in conservation (R→R, S→S, I→I) reduction (R→S, R→I, S→I) or complete disappearance of degeneracy. Although several examples of S- and I-degeneracy
were found without the aid of computer, systematic investigation was
performed by means of our reaction-generating program ARGENT-1. (This
program, also applicable to search for new types of nondegenerate processes,
is demonstrated at the conference.) Some of the results thus obtained are of
interest for chemists; the corresponding processes are considered in detail.
Among theoretical problems, the interrelationship between regularity and
self-inverseness and also problems of the actual existence for some R-, S-, and I-degenerate transformations are briefly
discussed. For example, for cyclic topology graphs with 4-12 vertices or
cubic topology graphs with 4-10 vertices, no examples of S- and R-degeneracy,
respectively, are detected. The search for a counterexample to another
problem (associated with degeneracy in charged systems) required as many as
201,748,864 edge-labeled cubic graphs to be constructed by ARGENT-1. |
Abstract: MATH/CHEM/COMP 2005,
|
|
THE
EASILY CALCULATED GEOMETRY DEVIATION INDICES
AND THEIR APPLICATION TO CHARACTERIZATION OF MOLECULAR SYMMETRY Serge S. Tratch and Nikolai S. Zefirov Department of
Chemistry, Moscow State University, Moscow 119992, Russia An interest
to quantification of many molecular characteristics (such as connectivity,
symmetry, chirality, etc) has been significantly increased during last
decades. The distance-based method, making it possible to calculate symmetry
measures for any finite point systems, was suggested by Zabrodsky et al.1
and then somewhat improved by Alikhanidi and Kuz'min2. In the both
approaches, symmetry measure is associated with some symmetry element and the
result obtained always depends on the actual disposition of this element in
3D space. In contrast, the computationally very simple distance-based
technique originated in this report can be applied to any individual symmetry
operation or to an arbitrary set of such operations; the operations in
question are represented by permutations of p points with known co-ordinates xi,
yi, zi, i =1 – p. More specifically, for some permutation π (converting
points i and j into π(i) and
π(j)), its inverse permutation
π-1 converts the pair (π-1 (i), π-1
(j)) into original pair (i, j) and this fact allows to
compare the known distance d(i,j) with the
average value calculated
for any given set of permulations π1,π2,...,πn.
The Geometry Deviation Index Δ (DGI) of the p-point system with respect to these n permutations can be then easily computed; the expressions for
average Δ' and normalized Δ'' indices are also shown below. Δ = In
practice, the point systems are formed from (all or only skeleton) atoms of
organic structures and from vertices of polygonal or polyhedral figures. The
sets of permutations needed for calculation of GDIs are typically
represented by symmetric groups Sp,
by automorphism groups of (unlabeled or labeled) graphs or configurations,
and by preselected subgroups or cosets of a given group. Some numerical
results obtained for spatial models of mathematical and molecular graphs are
finally discussed in the lecture. 1. H. Zabrodsky, S.
Peleg, D. Avnir, J. Am. Chem. Soc. 114 (1992) 7843; ibid 115 (1993) 8278. 2. S.
Alikhanidi, V.Kuz'min, Zh. Strukt. Khimii 39 (1998) 548; J. Mol. Model. 5 (1999) 116. |
 ,
,
Abstract: MATH/CHEM/COMP 2005,
|
|
MOLECULAR SIMILARITY IN 3D SPACE USING DISTANCE CONSTRAINS MATRIX Oleg Ursu and Mircea V. Diudea “Babes-Bolyai”
University, Faculty of Chemistry and Chemical Engineering, Arany Janos 11,
400028 Molecular similarity is
one of the most subjective concepts in Chemistry and can be defined in a
multitude of ways. Here we present a method for calculating the similarity
between pairs of chemical structures represented by 3D molecular graphs. The
method is based on a graph matching procedure using a maximum common subgraph
(MCS) detection algorithm to compute the exact degree and composition of
similarity. The proposed MCS algorithm involves the maximum clique detection
problem and it is adapted for 3D molecular graphs. Graph matching procedure,
used in our approach, accommodates the conformational flexibility by using
distance constrains matrix, encoding distance ranges between atoms, rather
than fixing the atom pair distances. The distance ranges are generated from
the initial guess, followed by successive optimizations, using triangle and
tetrangle bound smoothing algorithms from distance geometry. A set of
dopamine receptor antagonists is taken into consideration for testing the
proposed method; these compounds play a critical role in diseases such as
Parkinson’s and schizophrenia. Using the similarity
scores from 3D molecular similarity procedure, we proposed an ordering for
the studied compounds and compared them with the experimental biological
activities. The procedure can be used as a pruning in the preliminary stage
of rational drug design, in diseases involving dopamine neurotransmitters and
other classes of biologically active compounds. |
Abstract: MATH/CHEM/COMP 2005,
|
Multidimensional
approach to
|
Abstract: MATH/CHEM/COMP 2005,
|
|
Areas of some polygons and volumes of fullerenes Darko Veljan Department of Mathematics, Problems of
computing areas and volumes are among the oldest, hardest and most important
in mathematics and its applications. In principle, the answer to these questions
gave |
Abstract: MATH/CHEM/COMP 2005,
|
|
Deuterium
Isotope Effects in NMR Spectra Dražen Vikić-Topić Ruđer Bošković Institute, NMR Center,
P. O. Box 180, Zagreb, Croatia Deuterium
is often studied isotope due to the large fractional mass change on
deuteration as well as because of easy of deuterium incorporation into the
molecules. Deuterium substitution generates some peculiar effects into the
molecules: optical activity in CXYHD: small dipole moment (10-2-10-4
D) in monodeuterated methane, acetylene and benzene, which enables the
measuring of pure rotational spectra of these compounds. The most unusual
feature of deuterium labeling of organic compounds is the existence of long
range deuterium isotope effects on 13C NMR chemical shifts
(LRDIE). The high magnetic field NMR spectrometers enabled detection of LRDIE
even through 12 bonds, as small as 0.1 Hz, in p-electron systems. The
calculations of nuclear shileding changes1 due to LRDIE are still
challenging because of their low magnitude (3-300 ppb) and still not enough
developed theoretical approach for isotope interactions through many bonds. Generally,
isotope effects on chemical shifts are interpreted in terms of vibrational
and rotational averaging of nuclear shielding. Changes in nuclear shielding
with bond extension and/or bond angle deformation are introduced by
deuteration. In the theoretical model of LRDIE two contributions have to be
taken into account: (1) the secondary change in shielding at remote C-atom
due to shorter C-D than C-H mean bond length at the site of deuteration and
(2) the primary change in remote C-atom shielding, due to change in mean bond
length at this remote site. We assumed that LRDIE predominantly arise from
the change of bond length at the site of deuteration (1), since vibrational
differences at remote C-atoms of isotopomeric molecules are practiacally
undetectable. The C-D bond was modeled by reduction of the corresponding C-H
bond in the range 0.003-0.018 Å. Nuclear shieldings (GIAO) and atomic
charges (Löwdin, Mulliken) were calculated by different ab initio basis sets. For C-atoms more
than four bonds away from the deuteration site the differences of shielding
and charges between protonated and deuterated molecules correlate fairly good
with experimental LRDIE. By this approach experimental LRDIE can be
successfully calculated, even those over 10 and 12 C-C bonds. 1. D. Vikić-Topić, Lj. Pejov (2001) J. Chem. Inf. Comput. Sci. 41: 1478-1487. |
Abstract: MATH/CHEM/COMP 2005,
|
|
Kekulé Structures of C70 Damir Vukičević1 and 1Department of Mathematics, 2National The fullerene C70 is one
of the two most important fullerenes. We present the following properties of
this fullerene:
|
Abstract: MATH/CHEM/COMP 2005,
|
On generalization of the Hosoya-Wiener polynomial
Blaž Zmazek1,2 and Janez Žerovnik1,2 1FME, 2IMFM, Jadranska 19, SI-1000 The Hosoya-Wiener
polynomial1,2 of a graph G is defined as
Obviously, It is well known that the
(unweighted) Wiener number3 is the value of the first derivative
of the Hosoya-Wiener polynomial at x=1,
W(G) = W’(G;1). We can generalize the Hosoya-Wiener polynomial to weighted
graphs as follows. Let G be a edge and vertex weighted graph. WW(x)
is defined as
where d(u,v) is the graph distance between u and v. Clearly, the new
definition is equivalent to original definition if all vertex weights are equal
to 1. Note that WW(x) is in general not a polynomial if
arbitrary edge lengths are allowed. We will discuss algorithms
for efficient computation of WW(x) on trees and on cacti. Our results
include: Theorem: The Wiener polynomial on a vertex weighted tree T can be computed in O(D
Δ2 n) time, where D is the diameter of T and Δ is the maximal degree of a vertex in T. 15. H. Hosoya
(1988) On some counting polynomials in chemistry. Discrete Appl. Math. 19:
239-257. 16. B.E.Sagan,
Y.-N.Neh, P.Zhang (1996) The Wiener polynomial of a graph. Int. J. Quantum Chem. 60: 959-969. 17. H.Wiener (1947) Structural determination
of paraffin boiling points. J. Amer.
Chem. Soc. 69: 17-20. |
Abstract: MATH/CHEM/COMP 2005,
|
Exact quantum treatment of finite-dimensional system in the
interaction with the known
infinite‑dimensional system
Tomislav P.
Živković
Ruđer Bošković
Institute, P.O.B. 180, HR-10002 An important problem
in science is to describe properties of relatively small systems (atoms,
molecules or molecular fragments) that interact with surrounding media (other
atoms, molecules, molecular fragments, electromagnetic fields, crystals,
solute, etc.). Classical description of such systems is not sufficient, and
one has to use quantum theory. One can obtain relatively reliable quantum
description in two extreme cases. One extreme are small isolated quantum
systems Sa (such as
atoms and small molecules) which can be relatively well described (using
modern fast computers) with the standard Schrödinger equation. Other
extreme are some infinite quantum systems Sb (such as crystals – solid state,
electromagnetic field etc.) which have some special regularity such as translational
invariance. Utilizing this regularity one can obtain a relatively correct
quantum description of those infinite systems. Exact quantum description of
the combined system S=Sa |